CH 391: UNIT 3: STEREOCHEMISTRY
I.Stereochemistry of Nucleophilic
Substitution Reactions at Saturated Carbon
Nucleophilic
substitution at saturated carbon is a very common and useful reaction type.
These reactions are found to occur via two distinct mechanistic types, which are designated SN1
and SN2. The
stereochemistries associated with both of these mechanistic types will be
discussed, beginning with the SN2 reaction.
A.The Stereochemistry of SN2 Reactions. The
symbol SN2 refers to “substitution, nucleophilic,
bimolecular”. The
designation bimolecular refers to the number of molecules in the
rate-determining step. Generalized examples of SN2 reactions are
represented below, recognizing that the nucleophile may be negatively charged
or uncharged and the leaving group (when present in the substrate) may be
uncharged or positively charged.
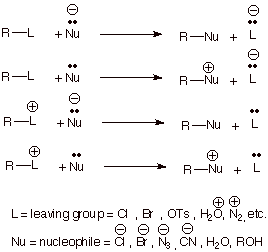
Whatever
the charge type of the nucleophile or the leaving group, these reactions
invariably occur with complete inversion of configuration at the saturated (sp3)
carbon undergoing substitution. One of the earliest (and especially elegant)
demonstrations of inversion of configuration in an SN2 reaction is
illustrated in the reaction of optically pure 2-iodooctane with radioactive
iodide ion. The observed reaction,t he incorporation of radioactive iodine into
the substrate, is rigorously second order (first order in iodide ion and first
order in iodooctane), as it should be for an SN2 mechanism. The key
observation is that the rate of incorporation of radioactive iodine into
2-iodooctane is exactly one-half
of the rate of racemization of the optically active substrate. This can only be
the case if each reaction of radioactive iodide ion with 2-iodooctane racemizes
two molecule of this substrate. In turn, this can only be the case if each
reaction occurs with inversion of configuration, the inverted product, combined
with a molecule of the original product, then provides two molecules of racemic
(R + S) 2-iodooctane. If the
reaction had occurred via a carbocation route, racemization would have been the
result of each reaction with radioactive iodide ion, making the rate of
racemization exactly equal to the rate of radioactive iodide ion incorporation.
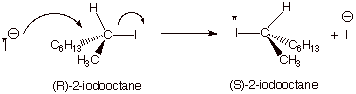
Of course the determination of absolute configurations of reactants and products now makes the determination of reaction stereochemistry relatively straightforward. The theoretical question of especial interest is why the preference for inversion over hypothetical retention is so powerful in all of these reactions. Incidentally, it clearly has nothing fundamentally to do with the circumstance that a negatively charged nucleophile may electrostatically repulse a leaving group, which in the molecule is uncharged but which is also in the process of becoming negatively charged in the transition state, in a retentive mechanism, since inversion is still strongly preferred even when the leaving group (when present in the molecule) is positively charged and the entering nucleophile is negatively charged (resulting in an electrostatic attraction). The theoretical baisis for the highly developed preference for an invertive reaction mechanism can be seen by applying either the concept of transition state aromaticity/antiaromaticity or by applying frontier orbital (HOMO/LUMO) theory. The former approach has already been developed, and for the specific case of nucleophilic substitution, in Unit 1. The explanation given there was that the transition state for retentive substitution is unavoidably a cyclic one and it possesses four electrons (two from the nucleophile and two from the C-L bond), so it is antiaromatic, whereas the transition state for the invertive process is acyclic, so that this TS is non-aromatic.
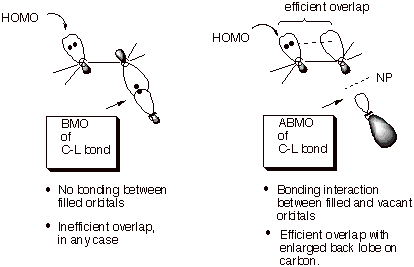
Let’s now consider the application of HOMO/LUMO (or FO) theory , which was also developed in Unit 1, to the specific case of bimolecular nuceophlic substitution. The electron pair being donated clearly must come from the nucleophile; so the HOMO is the orbital of the nuceophile (Nu) which contains the unshared electron pair used to bond to carbon. But what about the LUMO? Clearly, the substrate molecule must supply the LUMO. Since the bond being broken is the C-L bond, the appropriate LUMO must be that of this bond. Recalling that this antibonding LUMO of the C-L bond has a node in the center part of the bond, we can see from the illustration that whereas the nucleophile can bond to the carbon end of the bond, as it needs to, it cannot at the same time bond to the L part of the bond. The spatially unavoidable interaction with L is in fact antibonding, and tends to diminish the net bonding interaction of the nucleophile with the organic substrate. In contrast, if the nucleophile enters from the backside of the C-L bond, it is quite remove from L and there is no significant antibonding interaction to interfere with the bonding interaction between the nucleophile and carbon. It is important to realize that, although bonding interactions between the nucleophile orbital and both the carbon and L orbitals are possible if we use the C-L BMO, this interaction would be between two filled orbitals, and thus can yield no net stabilization. Remember that this is because in the interactions between two filled orbitals, there are four electrons to accommodate.
TS for the Hypothetical Retention Mechanism
B. The Stereochemistry of SN1 Reactions. The
generalized mechanism for an SN1 reaction is illustrated below. In
it the rds is heterolysis of the C-L bond, resulting in the formation of an
intermediate carbocation. The planarity of this latter ion typically results in
the its reaction, in approximately or exactly equal amounts from either face,
giving approximately equal amounts of retention and inversion, also described
as racemization.
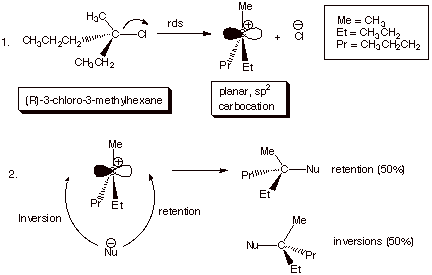
In a few cases, a slight excess of inversion over retention is observed (e.g., 90% racemization, 10% inversion), presumably as a result of weak “blocking” of the front face of the carbocation by the leaving group.
C. Retention
in SN Reactions.. In a
number of special cases, the stereochemical result of SN Reactions
is pure retention. These cases are
of special interest because neither of the typical SN
mechanisms provides for retention. One such reaction is the conversion of
3-bromo-2-butanol to 2,3-dibromobutane by reaction with hydrogen bromide. When
the (2R,3S) stereoisomer of this alcohol (the carbon atom bearing the hydroxyl
group is C2 and that bearing the bromo group is C3) is treated with aqueous
hydrogen bromide, meso-2,3-dibromobutane is produced diastereospecifically.
Note that the overall result is the replacement of the alcohol function by
bromine with retention of
configuration, since inversion (as in an SN2 mechanism) at the
alcohol carbon (C2)would have given the (2S,3S) stereoisomer of
2,3-dibromobutane (the configuration at C3 remaining fixed, of course). On the
other hand, reaction via an SN1 mechanism would have given a mixture
of 2S,3S and 2R,3S stereoisomers, i.e., a mixture of the meso diastereoisomer
and the optically active 2S,3S enantiomer. This follows from the fact that a carbocation mechanism
would have left the configuration at C3 unaltered, but given a mixture of the R
and S configurations at C2. The actual result, retention of configuration, can be clearly seen in the illustration
below by simply comparing the structures of the reactant and product in the
Newman projections provided.
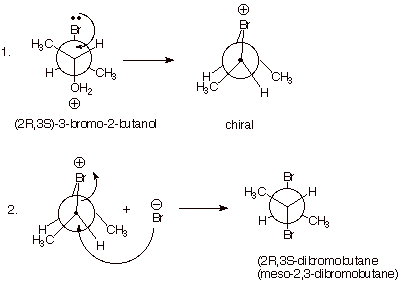
In
contrast, the (R,R) and (S,S) stereoisomers of the bromoalcohol both afford a
racemic mixture of (2R,3R) and (2S,3S)-dibromobutane. This amounts to retention
of configuration in the conversion of the (2R,3R) alcohol stereoisomer to the
(2R,3R) dibromide, but in inversion of configuration at both C2 and C3 for the conversion of the (2R,3R) alcohol
to the (2S,3S) dibromide. All of these results are readily and uniquely explicable
if it is assumed that bromine acts as a neighboring group (i.e., as an
intramolecular nucleophile), displacing the leaving water molecule from C2 with
inversion of configuration (as in a normal SN2 reaction) thus
forming an intermediate epibromonium ion. The latter then opens up by reaction
with the external nucleophile (bromide ion), as epibromonium ions do in what is
essentially an SN2 reaction, with inversion. Since the epibromonium
ion formed from either the (R,R) or (S,S) alcohol is achiral (it has a plane of
symmetry which bisects the central C2-C3 bond), it can react with bromide ion
at either carbon (the one which formerly was C3 or the one which formerly was
C2) equally to yield the two enantiomers of 2,3-dibromobutane. The distinct
epibromonium ions derived from the (2R,3S) and (2S,,3R) alcohol are chiral (do
not have a plane or other relevant element of symmetry), but their reaction
produces meso-2,3-dibromobutane, which is achiral. Thus, the products of all of
these reactions are optically inactive, even though they are formed from
enantiomerically pure starting alcohols.
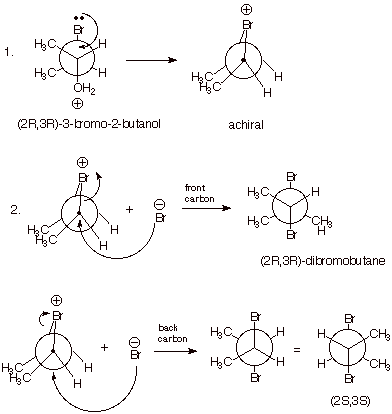
Analogous results are found in other reactions in which a suitably placed substituent having an unshared electron pair is present in the molecule (e.g., alkoxy, alkylthio, and amino, and amido substituents. Even certain electron-rich aryl substituents
(e.g. 4-methoxyphenyl = anisyl) are found to act as effective neighboring groups, e.g., in the solvolysis (cleavage of solvent) of the 3-anisyl-2-butyl tosylates, shown below. Note that the erythro/threo nomenclature for distinguishing diastereoisomers is convenient to use here. The erythro isomer of a compound containing two non-equivalent stereocenters is the one (like erythrose) in which all of the like groups can be placed anti to one another in one of the conformations accessible to the molecule. In the case under discussion, hydrogens are anti to hydrogens, methyls to methyls, and the tosylate (4-methylbenzenesulfonyloxy) and anisyl groups (the “like” groups which are not identical) are also anti. In the single product obtained in the acetolysis (solvolysis in acetic acid), the like groups are still anti to one another, i.e., only the erythro isomer is obtained.
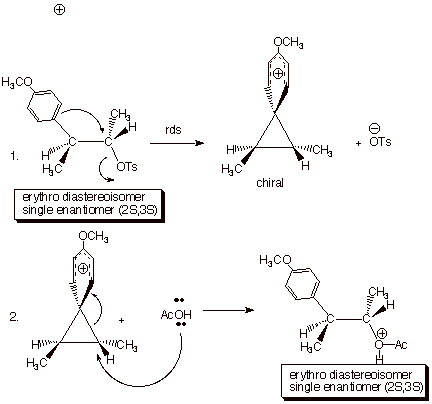
Further, only one enantiomer of the erythro diastereoisomer is obtained, since neither the intermediate arenium ion nor the product are achiral. In other words, both enantiomers of the erythro diastereoisomeric tosylates give different, enantiomerically pure products which are the result of retention of rigorous configuration at both stereocenters. In contrast, either threo enantiomerr gives an intermediate arenium ion which has a plane of symmetry and is therefore achiral. It generates racemic threo acetate (a 50:50 mixture of the 2R,3S and 2S,3R enantiomers). See if you can draw this out and show the structures of the two enantiomers of the threo product.Incidentally, the cationic intermediate in these reactions has been called a “phenonium ion”by Professor D.J.Cram, in whose research group this work was performed. It is a special kind of arenium ion, i.e., the type of cationic intermediate which is involved in electrophilic aromatic substitution. Finally, the participation of an internal nucleophile in the rds naturally results in rate enhancements in comparison to direct reactions with external (solvent) nucleophiles.
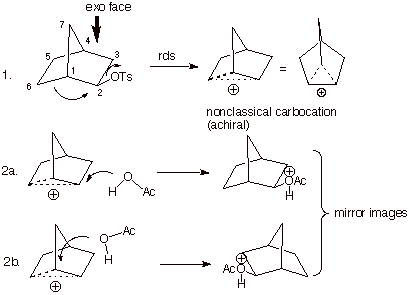
In
certain rigid bicyclic systems, such as the bicyclo[2.2.1]heptyl system (the
norbornyl system), even alkyl groups can act as neighboring groups. This is illustrated in the acetolysis
of optically pure exo- 2-norbornyl tosylate, which results in the formation
exclusively of the exo isomer, but in racemic form. Obviously, an SN2
reaction would result in the formation, exclusively, of the endo acetate,
whereas an SN1 reaction should result in the formation of a mixture
of endo and exo acetates. Importantly, in the case of either of these
mechanisms, an single enantiomer would result, i.e., optically active products
would be formed. Instead, only the exo product is obtained, and it is 100% racemic.
This results stem from neighboring group participation by the C6 carbon atom to
form a symmetrical (i.e., achiral), bridged cationic intermediate, which has
been called a nonclassical carbocation. The latter can react at either of two
equivalent carbon atoms (formerly C2 and C1) to yield both enantiomers of the
exo acetate in equal amounts.
II.
Stereochemistry of Additions to Alkenes
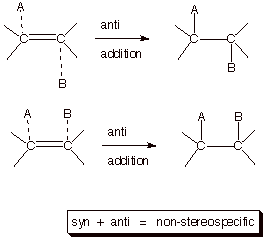
Additions to alkenes can proceed via three reqasonably distinct stereochemical outcomes,
viz., anti stereospecific, syn stereospecific, and non-stereospecific addition., as
indicated in the illustration given below. In anti stereospecific additions, the two new bonds to the
unsaturated carbon atoms of the alkene are formed from opposite faces of the pi
bond. In syn stereospecific
additions, the new bonds are formed exclusively from the same face of the pi
bond. Finally, in non-stereospecific addition, a mixture of syn and anti
addition occurs.
A. Additions
Involving Carbocations. Many additions to alkene pi bonds proceed via intermediate carbocations. In such cases the
stereochemical consequences are exactly the same as in SN1
reactions, viz., non-stereospecific reaction. Familiar instances are the
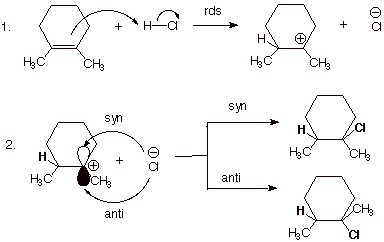
additon of HCl to alkenes and the acid-catalyzed hydration of alkenes. The
addition of hydrogen chloride to 1,2-dimethylcyclohexene is provided as an
illustration. The intermediate carbocation is able to react from either face of
the carbocation with near equal facility, giving both the cis and trans
products. Since the product chlorides are diastereoisomeric, the transition
states leading to them are diastereoisomeric, also, and thus have unequal
energies. Consequently, they are not formed in exactly equal amounts. However,
the amounts differ only slightly, since the reaction of the carbocation with
chloride ion (or water) is highly exothermic, providing for a transition state
which doesn’t have very much product character.
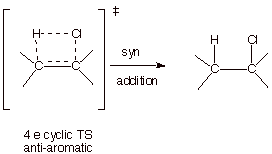
The question of why the reaction does not occur via a concerted, syn stereospecific path, instead of the
stepwise carbocation path is an interesting one. The carbocation path involves
a rate-determining step in which two bonds are broken and only one is formed.
It is strongly endothermic. On the other hand, the concerted addition pathway
would forrm two bonds and break two bonds and is obviously exothermic. Why is
the stepwise path, which appears to be less favorable from a thermodynamic
standpoint, actually the lower energy pathway. If we look at the TS for a
concerted addtion of HCl to a pi bond we can see that the TS has antiaromatic
character, i.e., it has a cyclic system which contains four electrons.
Consequently, the stepwise path is of lower energy. It should be noted that
concerted addition of HCl to 1,2-dimethylcyclohexene would give only the cis
chloride product (i.e., the product in which the methyl groups are cis o each
other on the cyclohexane ring).
Obviously,
acid-catalyzed hydrations of alkenes are similarly non-stereospecific, since
they also involve intermediate carbocations.
B. Anti Stereospecific
Additions to Alkenes. In sharp contrast to the acid-catalyzed hydration of
alkenes and hydrogen halide additions to alkenes, the additions of halogens to
alkenes occurs with rigorous anti
stereospecificity. This stereospecificity immediately notifies us that
intermediate carbocations are not involved. Nor is the addition likely to be
concerted, since concerted additions to alkenes should result in syn stereospecificity. Anti stereospecificity results from the initial addition of one
bromine (the electrophilic bromine) to the pi bond, followed by the opening of
this intermediate epibromonium ion from the opposite face by bromide ion. The
stereochemistry of this opening is exactly as expected, since it is essentially
an SN2 reaction, involving bromide ion as the nucleophile and the
bridging bromonium as the (internal) leaving group, which leaves from the face
opposite to that of the entering nucleophile. Recall that frontside
displacement of the leaving group (with the resulting syn stereospecific addition) would involve a cyclic, four
electron (antiaromatic) system. The addition of chlorine and iodine to alkene
pi bonds also occurs via an anti
stereospecific route.
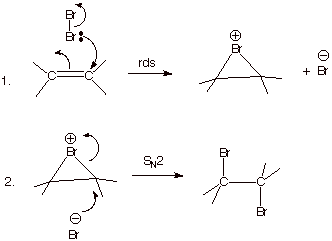
In
contrast, the addition of bromine (and other halogens) to alkenes in which the
prospective carbocation intermediate is stabilized by an electron donating or
conjugating group does not usually occur via an exclusively anti stereospecific route. Apparently, the initially
formed bromonium ion is able to
partially equilibrate with the relatively stable open carbocation, which
then can reclose, prior to reaction with bromide ion, to the diastereoisomeric
epibromonium ion, thus leading to a mixture of the two diastereoisomer
products. This is usually particular noticeable for the cis alkene because the epibromonium ion
corresponding to the cis alkene is
less stable (steric repulsions) than the trans diastereoisomer. One
reason the reaction is still considered to proceed initially via the
epibromonium ion is that trans-stilbene yields essentially only the meso
dibromo compound, via anti stereospecific addition.
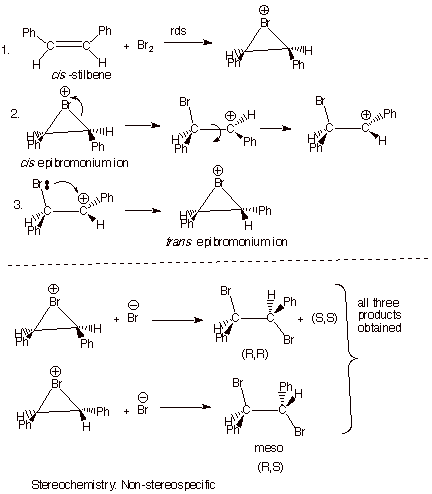
Regiochemistry of Bromine
Addition to Alkenes. In the bromination of alkenes, the two
bromine atoms introduced into the molecule enter into the mechanistic pathway
in entirely different roles. One enters electrophilically and the other as the
nucleophilic bromide ion. Unfortunately, these two bromines can not be
distinguished in the final product, so that even in additions to
unsymmetrically substituted alkenes, the regiochemical preference of
bromination cannot be directly determined. The question being explored here is
as follows: in the reaction of bromine with an unsaturated alkene such as
propene, does the nucleophile enter in selectively a the primary carbon or the
secondary carbon of the alkene or does it, in fact, enter unselectively. The
answer, which we can determine indirectly, but decisively, is that it enters
with a high degree of selectivity onto the more hindered, secondary carbon. The
conventional experimental determination uses the reaction of bromine with an
unsymmetrically substituted alkene in aqueous solution, where the same
epibromonium ion is formed as an intermediate, but this intermediate reacts
extensively with the more abundant water molecules to give a bromohydrin, i.e.,
a bromoalcohol. In the case of propene, as is illustrated below, the result is
selective formation of 1-bromo-2-propanol, rather than 2-bromo-1-propanol. It
is presumed that nucleophiles in general prefer this entry mode, and this is
supported by an analysis of the reasons for this manner and high degree of
regiospecificity. Incidentally, analogous results are obtained even in the
addition of aqueous bromine to a 1,1-disubstituted alkene such as isobutene,
where the competition is between a tertiary carbon and a primary carbon. The
nucleophile strongly prefers to react at the more highly substituted (more
sterically hindered!!) carbon.
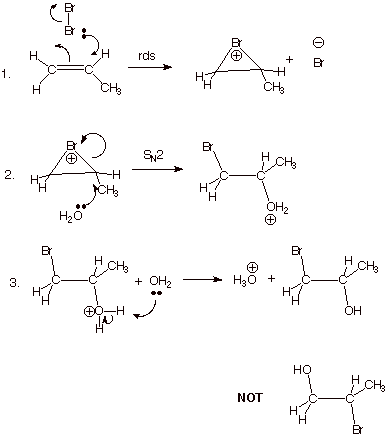
To
analyze the fundamental reasons for the somewhat surprising regiochemical
preference in the opening of the bromonium ion, we should first consider the
structure of the epibromonium ion intermediate. This is especially important
because the second step fo the reaction, that in which the epibromonium ion is
opened by a nucleophile, is highly
exergonic (the epibromonium ion is not so much more stable than an analogous
carbocation). Consequently, an applicationi of the Hammond Principle indicates
that the transition state for this reaction step should more closely resemble
the reactant in this step, which is the epibromonium ion. In general, the
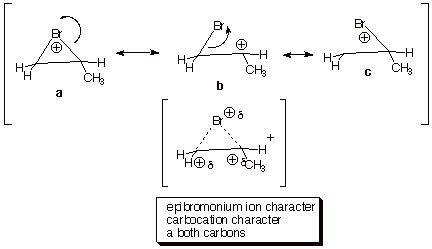
cationic species which we call an epibromonium ions has three reasonable
resonance (canonical) structures. Only one of these is a bromonium ion
structure (see below), while two carbocation structures also contribute to the
resonance hybride. Consequently, the epibromonium ion has both bromonium ion
character and carbocation character at both carbons of the original double
bond. In the case of an unsymmetrically substituted alkene such as propene, the
two canonical carbocation structures are not of equal energy, and resonance
theory requires that the hybrid (real) species more closely resemble the lower
energy structure. In the resonance treatment of the epibromonium ion derived
from propene, the structure labeled a
has positive charge at a secondary carbon. It is therefore of lower energy than
the structure c, which has
positive charge at a primary carbon. The real, hybrid species therefore more
closely resemble structure b, so
there is more positive charge at the secondary carbon than at the primary
carbon. The short form of the rationale for the attack of the nucleophile at
this carbon is that ions typically react at the site of highest charge density.
This is expected because the reaction of an epibromonium ion with a nucleophile
is exothermic, so that the TS of the second step of the reaction also resembles
the epibromonium ion (rather than the product of that step). This implies that
covalent bond formation hasn’t proceeded very far, so that the
nucleophile-to-carbon bond is still long and largely ionic. There are two
important insights from this picture. First, the knowledge that a long and weak
covalent bond exists in the TS suggests that steric effects should be quite
small. Thus the distinction between tertiary, secondary, and primary carbons on
the basis of steric effects (which consideration is dominant in SN2
reactions) is at most slight. Secondly, the ionic (or electrostatic) nature of
the bonding will be strongest when the fractional positive charge is largest,
which it is on the secondary carbon in the present instance. It is interesting
to note that these regiochemical considerations remain valid even when the
choice is between a tertary and a primary carbon, as in the case of the reaction
of isobutene with bromine in aqueous or alcoholic solution.
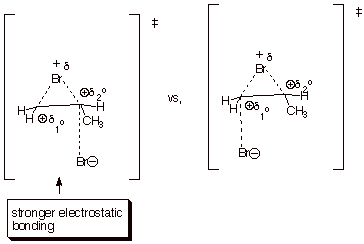
Invoking
the Method of Competing Transition States, as illustrated below, the TS for
reaction of the nucleophile at the secondary carbon is favored because:
q
The reaction step is exothermic,
so the TS closely resembles the epibromonium ion plus bromide ion. Consequently
the covalent bonding between the bromide ion and the appropriate carbon is weak
and long in the TS. As a result, steric effects, as between secondary and
primary carbons are minimal.
q
Since the TS is much
like the epibromonim ion, and the covalent bonding is weak, the main source of
bonding interaction between the nucleophile and the appropriate carbon of the
epibromonium is is ionic or electrostatic bonding. Such bonding should be
stronger the larger the fractional positive charge on the carbon atom. Thus the
interaction with the larger partial positive charge on the secondary carbon is
stronger than that with the smaller partial positive charge on the primary
carbon.
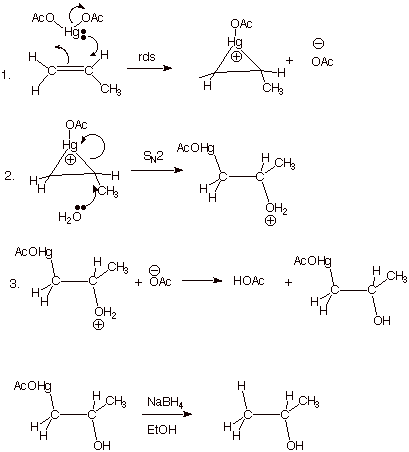
Oxymercuration.
Another alkene addition which
proceeds with anti
stereospecificity ( in the case of simple alkenes), is oxymercuration. The
addition of mercuric acetate to alkenes in aqueous solution proceeds to yield a
b-acetoxymercuri alcohol. The latter can be reduced to
the simple alcohol using sodium borohydride. The overall result of
oxymercuration/reduction is anti
stereospecific, Markovnikov (regiochemistry) hydration of an alkene (without
encountering carbocation rearrangements). The latter regiochemical feature is
illustrated below in the oxymercuration of the unsymmetrically substituted
alkene, propene. A further
advantage of this indirect hydration method is that skeletal and hydride
rearrangements do not occur, because carbocation intermediates are not
involved. As in the case of bromination, the anti stereospecificity is the result of the opening of a bridged
mercurinium ion from the opposite face in a nucleophlic substitution reaction.
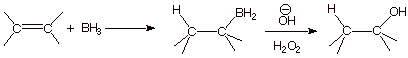
Syn Stereospecific
(Concerted) Additions. Additions to alkenes which proceed with syn
stereospecificity are nearly
always concerted additions. A familiar example is hydroboration, that
is, the reaction of an alkene with borane to yield an organoborane. Oxidation
of the organoborane in situ with alkaline hydrogen peroxide yields
an alcohol, with net syn stereospecific addition of water to the double bond,
without the troublesome rearrangements which accompany carbocation mediated
reactions, and with opposite regiochemistry (anti-Markovnikov) to that observed
in acid-catalyzed hydration.
The
resonance treatment of the transition state for this reaction has already been
given in Unit 1, but is repeated here. Note that in the TS the hydrogen which
is being transferred to carbon from boron is still partially linked to the
boron atom. Consequently, the formation of the new C-H bond and the new C-B
bond must take place from the same face of the pi bond. It would be
geometrically impossible for the hydrogen to be transferred to the opposite
face of the pi bond while still maintaining significant bonding to boron.
Essentially, this is why syn stereospecific reactions are almost always
concerted reactions and conversely, i.e., concerted mechanisms essentially
always result in syn stereospecific addition.
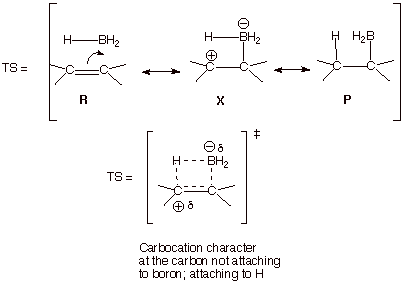
There
is another interesting aspect of this hydroboration reaction. Since we seem to
have a cyclic TS with four electrons (two from the pi bond and two from the B-H
bond), this would seem to set up
an antiaromatic TS, which should be strongly disfavored. In fact, this TS is
not rigorously cyclic when viewed in terms of the orbital overlaps. In
particular, the 2p AO on the carbon which is bonding to boron is bonding to a
2p AO on boron, but the hydrogen which is being transferred to carbon is bonded
to boron via another AO (a boron sp2 AO) which is orthogonal to (has
zero overlap with) the 2p AO on boron. Consequently, the overlaps of the system do not continue through
boron, which acts as an insulator between the hydrogen and the carbon to which
boron is bonding. Consequently, it should be recalled that not all
four-membering ring transition states necessarily are antiaromatic.
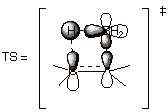
Carbene
Additions. Carbenes are species containing divalent carbon. The
addition of the simplest carbene (CH2also called methylene) or
substituted carbenes to alkenes is an interesting and preparatively useful
reaction. Dichlorocarbene (CCl2) is an especially readily available
carbene which, although not isolably stable, can be conveniently generated in
situ for preparative purposes. Since
the divalent carbon is uncharged and has only two covalent bonds, there are two
additional valence shell electrons to be distributed between the remaining two
valence shell orbitals. They can both go into the same AO with spins paired
(singlet carbene) or into different AO’s with unpaired spins (triplet
carbene). Although the ground state of the parent carbene is a triplet, both
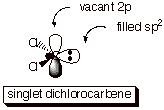
states can be accessed, depending upon the mode of preparation. The ground
state of dichlorocarbene, however], is a singlet. The unshared pair of
electrons occupies a hybrid orbital, leaving the higher energy 2p AO
vacant. The additions of this
substituted
carbene to alkene pi bonds,
as noted earlier, have been studied and used extensively. The addition, which
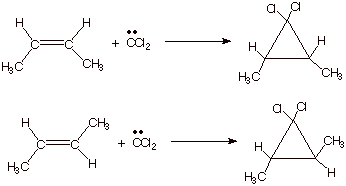
forms two new carbon-carbon bonds, while breaking none, is highly syn
stereospecific. For example, the addition to cis- and trans-2-butene
afford, respectively, the cis- and
trans cyclopropanes, as shown
below.
The rigorous syn stereospecificity suggests that the reaction is concerted, and theoretical calculations strongly support this conclusion. There are several unique aspects of this reaction path which deserve our attention. First of all, the reaction is an example of a relatively rare class in which the enthalpy of activation appears to be nil or vanishingly small. Entropy effects, however, do appear to play some role in defining the modest selectivity of carbene additions. The second novel aspect of this cycloaddition reaction is that it takes place via a non-least motion pathway in which the orientation of the carbene with respect to the alkene is drastically different from its orientation in the final, cyclopropane product. This is because the approach of the carbene to the alkene in a geometry which closely resembles that of the product would contain an antiaromatic 4 electron cyclic array. In contrast, theun-productlike approach has a two electron, aromatic cyclic array. Consequently, in the least energy pathway the carbene fragment must, somewhere along the reaction path, rotate a full 90 degrees to provide the ground state geometry of the cyclopropane product. This rather extreme motion is, however, performed subsequent to the transition state and after rather strong bonding has already been initiated, so that it is not problematic in regard to activation energy.
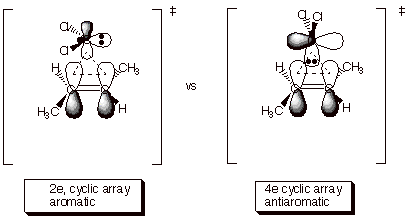
Since the carbene fragement has two AO’s, there is actually a second cyclic array present in either pathway. For example in the disfavored path (the one shown on the right above) there is also another cyclic array, one involving the vacant 2p AO (see below). Since the pi bond contains 2 electrons, this constitutes a 2 electron cyclic array, and would seem to represent an aromatic array. Quite the opposite is true, however. The array which involves the 2p AO is what is known as a Mobius array. A Mobius cyclic system is one which has one or an uneven number of nodes in the most bonding MO. The magic number for aromaticity in such arrays is just the opposite as for a Huckel array, i.e., 4n pi systems are aromatic and 4n+2 systems are antiaromatic. So it can also be seen that the second cyclic array in the favored path, which involves the doubly occupied sp2 AO of the carbene, is a 4 electron Mobius array, which is aromatic. Consequently, the favored path has two aromatic arrays and the disfavored one two antiaromatic arrays.
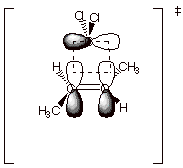
Diels-Alder
Cycloadditions. Perhaps the most famous, and also
the most useful, of all cycloaddition reactions is the Diels-Alder reaction of
a dienophile with the s-cis conformation
of a conjugated diene. Again, the reaction is highly syn stereospecific, both
with respect to the dienophile and the diene. The stereospecific reactions of
diethyl maleate ( which has cis ester functions, E) and diethyl fumarate
(trans E’s) with
1,3-butadiene are depicted below (NP = nodal plane). In the specific
depictions, the dienophile is shown as approached the termini (C1 and C4) of
the s-cis conformation of the
diene from above, so that the bottom lobes of the dienophile orbitals overlap
with the top lobes of the diene orbitals. The orbital signs suggested by the
light and dark lobes imply the interaction of the dienophile HOMO (which is
symmetric, like that of the BMO of ethene) with the LUMO of the conjugated
diene (this MO is also symmetrical and has two nodal planes, one between C1-C2
of the diene and one between C3-C4 of the diene). Note also that the
coefficients of the diene LUMO are larger the termini (C1 and C4) than at the
internal positions, as implied by the sizes of the AO’s depicted. Most
importantly, from the point of view of FO theory, the interactions between both
termini of the diene and the dienophile are bonding, a circumstance which
follows from the matched symmetrics of the dienophile HOMO and the diene LUMO.
The same favorable bonding situation is found when examining the interactions
of the diene HOMO and the dienophile LUMO (try this as an exercise).
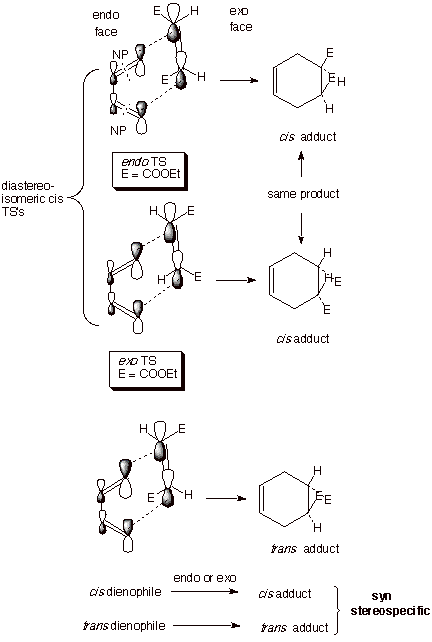
It
is also of interest to note that for the cis
dienophile there are two diastereoisomeric transition states, both of
which lead to the same cis
Diels-Alder adduct. The endo face of the TS is considered to be the one which
lies directly (or nearly directly) over the diene (especially C2 and C3), while
the exo face is the one which
projects away from this diene moiety. In one cis TS both ester functions are endo and in the other
both ester functions are exo. In the single TS for the addition of the trans dienophile, one ester function is endo and the other
exo. We shall see momentarily the reasons for the importance of recognizing the
endo and exo faces of a Diels-Alder transition state.
Not
only is the addition to the dienophile syn stereospecific but, as the drawing
implies, it is also a syn stereospecific addition to the termini of the
conjugated diene, i.e., both new bonds to the dienophile are formed from the
same face, here shown to be the top face of the diene. The consequence of this
stereospecificity element is that different geometric isomers of the diene lead
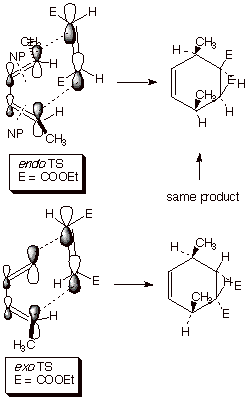
to different adducts. For example, addition to E,E-2,4-hexadiene leads to
adducts in which the two methyl groups are cis to one another in the adduct, as shown below. Again,
the cis dienophile can orient the
ester substitutents either endo or exo, but this time the adducts are
differentiated, and the endo one is slightly favored (more about this later).
The key points here are (1)the E methyl groups turn out to be cis to one another in the adduct and (2) the majority
endo product has the ester groups cis
to the methyl groups.
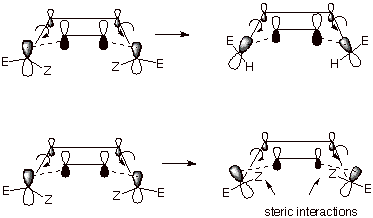
The illustration below indicates why the two E methyl substituents end up being cis to one another and cis to the endo substituent (or endo H, as the case may be). Specifically, the terminal carbons (C1 and C4) of the conjugated diene rotate the E groups toward the face of the entering dienophile and their Z groups away from that face in order to optimize overlap with the dienophile pi orbitals. This is necessary because the dienophile sits inside the perimeter of the diene system, so that the 2p lobes on the same face as the dienophile must be rotated inward, toward the dienophile pi system in order to maintain efficient overlap. The opposite sense of rotation (shown in the bottom diagram) would not only result in sharply diminished overlap between the diene termini and the dienophile pi orbitals, but would introduce strong steric interactions between the Z groups (even if these are only hydrogen) and the dienophile.
Stereochemistry of Beta Elimination Reactions
Elimination reactions are the reverse of additions,
and it has previously been noted that there are three potential stereochemical
outcomes in additions, viz., anti stereospecificity, syn stereospecificity, and
nonstereospecificity. Not surprisingly, the same three potential outcomes are
possible in eliminations. In this case we are speaking of the removal of two
atoms or groups (normally a beta hydrogen and a leaving group) from opposite
faces of the developing pi bond, fromthe same face, or a mixture of the
two. When the elimination
mechanism is a stepwise one (e.g., E1CB ), the stereochemical result
is usually nonstereospecificity. However, when the reaction is concerted, as in
the E2 elimination mechanism, the stereochemical preference is typically anti
stereospecificity. There appear to be two primary reasons for this preference
anti over syn stereospecificity. First, the anti elimination can proceed via a
TS in which eclipsing interactions are absent, whereas in the syn TS, eclipsing
or partial eclipsing occurs in the TS. A second possible reason, however, can
be given in terms of a HOMO/LUMO approach to the interaction of the developing
carbanion center at the beta carbon with the C-L bond involving the leaving
group (L). Since the carbanion center has a filled orbital, it must interact
with a vacant C-L orbital if there is to be a bonding interaction. Of course,
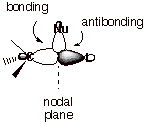
that would be the antibonding MO corresponding to the C-L bond. It has been
noted that, ion contrast to the BMO, the ABMO has an enlarged back lobe on
carbon. This is the result of the interference between the oppositely signed
AO’s on carbon and the leaving group in the ABMO. In effect, the orbital
density is “chased” from the antibonding region onto the back lobe.
The enlarged back lobe then overlaps more efficiently with the carbanion
orbital than does the diminished front lobe.