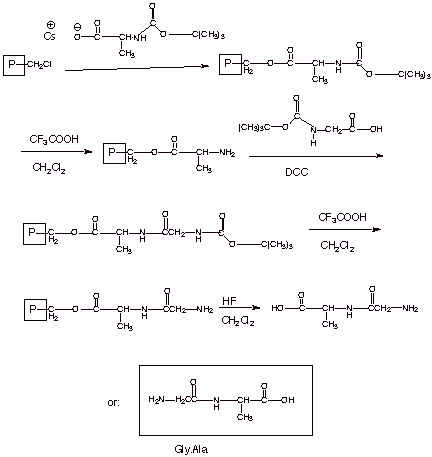CH 610B
Final Exam
Spring,2002
Friday, May 10
Prof.N.L.Bauld
Answer
Key
__________________________________________ Name (Last,
First)
__________________________________________ Social Security Number
Question |
Possible Points |
Page/Pts Earned |
Score |
|
I. |
37 |
2/11/ 3/9/ 4/7/ 5/4/ 6/6/ |
|
|
II. |
37 |
7/11/ 8/7/ 9/7/ 10/12/ |
|
|
III. |
29 |
11/9/ 12/10/ 13/10/ |
|
|
IV. |
20 |
14/10/ 15/10/ |
|
|
V. |
29 |
16/7 17/11 18/11 |
|
|
Totals |
152 |
|
|
I. Conjugated
Dienes
A.Structure and Nomenclature
1.
[2 pts] Provide the IUPAC name and
numbering system for the following conjugated diene:
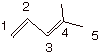
4-methyl-1,3-pentadiene
2.
[3 pts] Draw Newman projection
structures for both planar
conformations of 1,3-butadiene and
provide the appropriate names for
them.
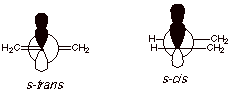
3. [2 pts] Using the above drawings, explain why these planar conformations are strongly favored over other, nonplanar, conformations. Show any relevant overlaps by means of orbital depictions.
Only in the planar conformations are the 2p AO’s at C2 and C3 parallel, so as to give maximum overlap and resonance stabilization. (See drawing above for orbital overlaps)
4.
[1 pts]Name one type of effect would normally tend to disfavor the planar
forms. Torsional effects.
5.
[1 pts] Which one of the two planar forms is the more stable? Explain, using a
structural illustration. The s-trans-conformation is the more stable one
because of the inside H/H steric repulsion in the s-cis conformation.
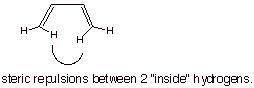
6.
[2 pts] The diene the structure of which is shown in question 1 above does not
undergo the Diels-Alder reaction at all. Explain this total lack of reactivity, being very specific about the
structural effect you are proposing and illustrating it.(Hint: Draw the s-cis conformation and examine it) The s-cis
conformation is required to undergo the Diels-Alder reaction, but cannot be
readily formed by this diene because it has an inside methyl group, which
exerts a powerful steric repulsion vs. the inside H.
![]()
7. [1 pts] Show the Newman projection structure of the conformation of 1,3-butadiene generated after a rotation of 90 degrees from either planar conformation. How much resonance stabilization is present in this conformation? Explain, again using orbital depictions.

B. Conjugate Addition
1.Mechanism
of the Addition of HBr to 1,3-butadiene.
a.
[3 pts]Write the detailed mechanism for the addition of hydrogen bromide to
1,3-butadiene as carried out at –78 degrees. Draw the structure of the
products and indicate which product is major. [route b is the major route]
![]()
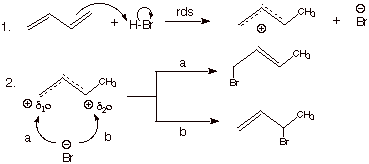
b.
[3 pts] Provide a resonance theoretical treatment of the intermediate
carbocation and summarize as a dotted line/partial charge structure.
Characterize this structure as completely as you can, indicating at which
carbon the positive charge is largest and explaining why in terms of resonance
theory.
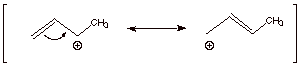
Resonance Theoretical
Treatment
Dotted Line/Partial Charge
Structure
![]()
q
The first structure
drawn above is of lower energy than the second canonical structure, because the
first is a secondary carbocation, while the second is a primary carbocation.
q
The real structure
(resonance hybride) more closely resembles the lower energy structure, which
has a secondary carbocation center.
q
Therefore, the
resonance hybrid has more carbocation character at the secondary than at the
primary carbon.
c.
[2 pts]Use this characterization to explain the predominance of one product
over the other. Make your argument stepwise and logical.
q
The bromide ion
reacts preferentially at the site of highest positive charge density.
q
That is, at the
secondary carbon, to give:
![]()
d. [1 pt]What kind of control is this?
q
This is kinetic
control (or, rate control).
2.
Addition of HBr to 1,3-Butadiene at Room Temperature
a. [1 pts]
Indicate which product is the major one at room temperature, and indicate what
kind of control is involved in these circumstances.
q
Thermodynamic (or,
equilibrium) Control.
q
This gives the major
product:
![]()
b. [2 pts] Provide a mechanistic explanation of how these
two products are interconverted.
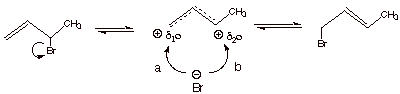
c. [1
pts]Provide a specific structure-based explanation of why the major
product is the favored one under these conditions.
q
This is a
disubstituted alkene, whereas the other product is a monosubstituted alkene.
q
Disubstituted alkenes
are more stable than monosubstitued ones (alkyl substitution stabilizes double
bonds.
3.
Thermodynamic vs. Kinetic Control
a.
[2 pts] Suppose two products, A and B, are formed in a certain reaction and
product A is the major product. You want to know whether this product mixture
is kinetically controlled or thermodynamically controlled. Explain how you
could decisively determine what kind of control is operative.
q
Purify A and B
q
Subject each
individually pure isomer to the exact reaction conditions, including
temperature, solvent, reaction time, etc.
q
If A remains pure A,
without isomerizing, and B remains pure B, after this reaction time, the
product mixture was kinetically controlled.
q
On the ther hand, if
pure A and pure B both rearrange
to the same mixture of A and B as obtained in the original reaction, the
product mixture is thermodynamically controlled.
C. Diels-Alder Cycloaddition
1.
[3 pts] Consider the prototype Diels-Alder cycloaddition reaction between
1,3-butadiene and ethane, shown below. Using resonance theory, derive a TS
model for this reaction and summarize as a dotted line structure. Characterize
the TS model as carefully as you can. Use this TS model characterization to
explain why the Diels-Alder reaction is so facile (easy and fast).
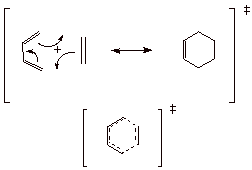
q
Cyclic 6e conjugated
system
q
Aromatic TS
3.
[1 pts] Consider the TS model for the reaction between two ethene molecules to
give cyclobutane. Characterize this TS model in the same way as for the
Diels-Alder reaction, and explain why this reaction is not observed. (Of course,
ring strain in the cyclobutane ring is also a factor, but do not consider ring
strain in your answer).
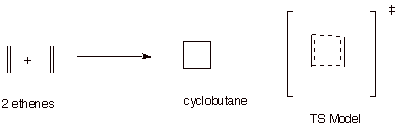
q
This has a cyclic
conjugated system of 4e.
q
This system is
antiaromatic (or non-aromatic)
4. [2 pts] Draw the
structures of the Diels-Alder adducts obtained in each of the following
reactions.
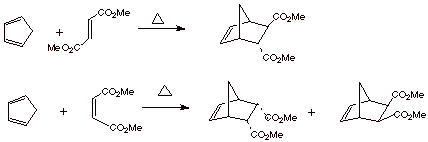
5.[
2 pts each = 4 pts] Provide the appropriate numbering systems and derive the
IUPAC names of the following bicyclic molecules.
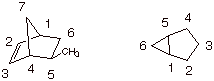
q
The first is:
exo-5-methylbicyclo[2.2.1]-heptene-2 {the exo will not be required, but give 1
extra point if it is present}
q
The second is:
bicyclo[3.1.0]hexane.
II. Polymerization
A.. Radical Chain Polymerization
1.
[3 pts] Mechanism of the Polymerization of Vinyl
Chloride (Chloroethene).
a. Write the detailed mechanism for the radical
polymerization of vinyl chloride.
You need not write the termination steps, but carry the propagation
steps out to the point where three monomer units are incorporated.
![]()
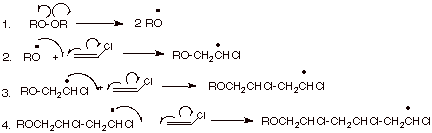
b.
[2 pts] What kind of initiator is usually employed (draw a structure and
provide a generic name)? Explain why these compounds are effective initiators.
q
A peroxide ( or, an
organic peroxide), as shown above.
q
The O-O bond is
unusually weak, and is readily homolyzed at convenient temperatures.
c.
[2 pts] Draw the repeat structure of the polymer and provide its formal name.
![]()
q
The formal name is:
poly(vinyl chloride) ; note that the parentheses are necessary because the
monomer name is compound.
d. [4 pts] Consider the three possible structures of the
polymer of vinyl chloride given below. In what general kind of selectivity do they differ from each other? Indicate the specific
names given to each polymer structure according to the sub-type of selectivity involved. Finally, which type of polymer is actually
observed in the polymerization of
vinyl chloride?
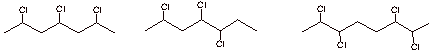
A B C
q
They differ in regiospecificity
or regioselectivity.
q
A is head-to tail
regiospecific; B is non-regiospecific; C is head-to-head regiospecific.
q
A is favored.
e. [2 pts] Based upon the two TS models given below for
two different selectivity modes, explain the specific selectivity preference
observed with vinyl chloride, based upon comparative characters of the two
TS’s. (Do not consider steric effects).
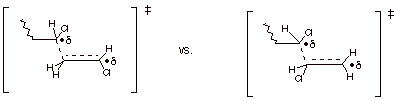
This has radical character This
has primary radical character
stabilized by a chlorine
substituent
It is therefore preferred
over primary
radical character.
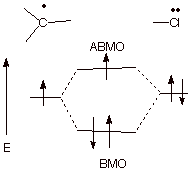
f. [2 pts] Draw a molecular orbital diagram which illustrates the effect a chlorine substituent
has upon a radical site and indicate the specific name given to this type of bonding interaction.
![]()
q
This is known as 3
electron bonding.
2.
Polymerization of Ethene.
a. [3 pts] Provide a mechanistic explanation, using
appropriate structural illustrations, of why the polyethylene obtained in the
radical polymerization of ethane is not of the “straight chain”,
i.e., unbranched variety.
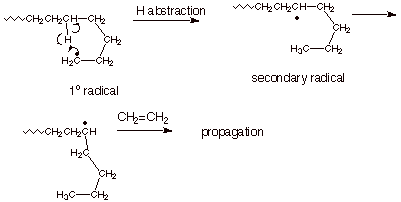
b. [1 pts] Explain why, in contrast, the polymer derived
from vinyl chloride by radical polymerization is not branched.
q
In this case, the
radical is not primary , being stabilized by the chlorine substituent, so the
radical is more stable and not as reactive toward hydrogen abstraction.
B. Chain Growth Polymerization
1. Nylon 66
a. [1 pts] Illustrate, by equation, what product is
formed initially when the two components of Nylon 66 are combined at room
temperature
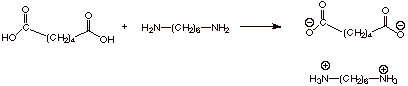
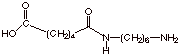
b. [2 pts] Upon heating this initial product,
polymerization begins. Draw the structure of the first product, loosely
speaking a “dimer”, which is formed in this reaction. With what
kind of bond are the two monomers attached together? Amide bond.
c. [2 pts] Illustrate the structure of
“trimer” which would be formed in the next step.
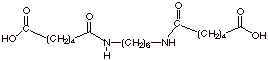
(This is one trimer; another
one is also possible)
d. [1 pts] Provide the “repeat structure” of
Nylon 66. Why is it called “66”? Both comonomers have 6 carbons.
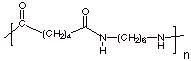
[Note to graders: There are a
few other correct ways of writing this]
C. Chain Growth vs. Step Growth Polymerization.
1. [1 pts] Indicate whether the polymerization of ethene
is a chain growth or step growth process.
Chain Growth
2. [1 pts]What kind of growth is involved in the Nylon 66
polymerization?
Step Growth
3. [2 pts] Specifically indicate an experimental test for which type of polymer growth is involved in
a given polymerization reaction.
q
Stop the
polymerization after short reaction times (or, low monomer conversions, such as
5-10%).
q
If the monomer
consumed is converted to high polymer, this is a chain growth process.
q
If only dimers,
trimers, or low oligomers are formed, this is a step growth process.
q
D. Ziegler-Natta Polymerization
1.Mechanism
of the Ziegler-Natta Polymerization of Ethene
a. [3 pts] Sketch the mechanism for the Ziegler-Natta polymerization of 1,3-butadiene
and provide the specific structure of this polymer.
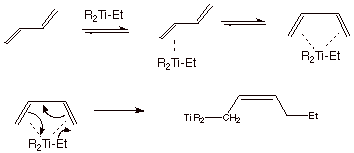
Structure:
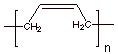
b. [2 pts]What is unique about the structure of this
polymer (Two different selectivity elements).
Selectivie
1,4-addition to the diene (conjugate addition)
Selective
addition to the less prevalent s-cis conformation to give the the cis- polymer.
[1
pt]What kind of properties does this polymer have?
q
It is elastomeric
(rubbery).
E. Stereoregular Polymers
1. [2 pts]Indicate the specific names associated with the
stereoregular polymers shown below.
![]()
isotactic syndiotactic
III.Carbohydrates
A. Classifications, Fischer Structures, and Stereoisomerism
1.
[1 pts] The structure of D-erythrose is provided
below. Draw the Fischer structure of its enantiomer and explain why the
structure provided is classified as the D enantiomer.
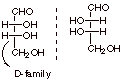
q
The D family has the
OH group to the right at the stereocenter most remote from the aldehyde
functionality.
2.
[3 pts] Draw the Fischer structures of any other stereoisomers of D-erythrose
and classify them as D or L. How many stereoisomers are there, counting both D-
and L-erythrose and all of their stereoisomers? Explain why this is this
expected number of stereoisomers based upon the number and type
of stereocenters and a specific rule.
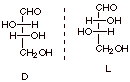
q
There are 4
stereoisomers
q
There are two
stereocenters
q
The 2n Rule.
3.
[2 pts] Provide the Fischer structures and names of two aldopentoses (including
whether they are D or L).
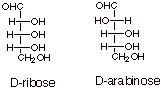
4.
[2 pts] Draw the Fischer structures of D-glucose and D-mannose.
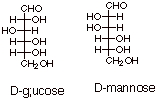
5.
[1 pts]Draw the Fischer structure of L-glucose. Which is more thermodynamically
stable, D- or L-glucose. Explain.
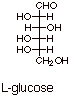
q
They are both equally
stable, since they are enantiomers.
B. Cyclic Structures
1.
[3 pts] Draw the conformational (chair) structures of the two
cyclized forms of D-glucose and provide the complete name of each. What is the general name given to two isomers of this type?
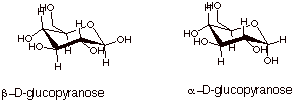
q
These are called anomers.
2.
[1 pt] Indicate which one of the cyclized structures is more stable and why.
q
The beta anomer is
more stable because the hydroxyl group at C1 is equatorial, whereas the
hydroxyl group at C1 in the alpha anomer is axial.
3. [1 pts] Draw the conformational structure of the two cyclized forms of D-mannose. Compare the thermodynamic stabilities of the more stable of these two cyclized forms of mannose with the more stable cyclized isomer of D-glucose. Which is more stable and why?
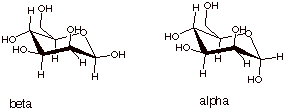
q
Even the more stable
beta anomer of mannose has one hydroxyl group axial, that at C2, while the beta
anomer of D-glucose has all hydroxyl groups equatorial.
q
So, the beta anomer
of the cyclized form of D-glucose is more stable than the beta anomer of the
cyclized form of D-mannose.
4.
[1 pt] Briefly explain, without any mechanistic sketch, why D-glucose and
D-mannose are relatively easy to interconvert..
q
They differ only in
their configurations at C2, which is an alpha type carbon with acidic alpha
type protons, which are easily removed to form the enol or enolate.
5.
[2 pts] Provide a mechanistic sketch of how the two cyclic isomers of D-glucose
are interconverted. What does this have to do with mutarotation?
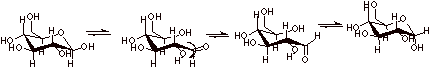
q
The equilibration of
alpha and beta anomers is what causes the change in rotation.
6.
[2 pts]Which is more stable, the acyclic (open chain) form or the cyclized form
of D-glucose? Explain this, making a comparison to the analogous reaction of an
alcohol with an aldehyde.
q
The cyclized form is
more stable, in contrast to the simple reaction of an alcohol with an aldehyde.
q
The intramolecular
hemiacetal formation is much more favorable than intermolecular hemiacetal
formation because the latter involves the combination of two molecues into one—i.e.,
the loss of translational entropy.
D. Disaccharides and
Polysaccharides
1.
[2 pts] Draw the conformational structures of cellobiose and maltose.
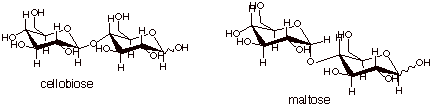
q
Note that either
anomer can be counted correct for either disaccharide.
2.
[1 pts] Indicate which one of these is more stable and explain why.
q
Cellobiose is more
stable, because it has a beta (equatorial) glycoside (acetal) linkage.
3. [2 pts] For cellobiose, indicate any functionalities which are of the glycoside or hemiacetal type and designate each one. Is cellobiose a reducing sugar? Why or why not?
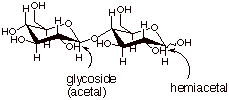
q
Yes, it is a reducing
sugar, since it has a hemiacetal function, which can equilibrate with the free
aldehyde form.
4.
[2 pts] For D-sucrose, the
structure of which is shown below, indicate any linkages which are of the
glycosidic and the hemiacetal type. Is this a reducing sugar? Why or why not?
Provide the correct numbering system for each ring.
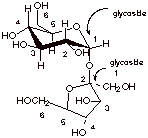
q
Since this sugar has
no hemiacetal linkages, it cannot equilibrate with the free aldehyde form in
neutral or alkaline solution, so it is not a reducing sugar.
5.
[1 pts] In general terms, explain how two monosaccharides can be linked
together in such a way as to give a non-reducing sugar which does not undergo
mutarotation.
q
The hemiacetal carbons
of both must be linked. (Or, the hydroxyl group of the hemiacetal carbon of one monosaccharide is linked to the
hemiacetal carbon of the other monosaccharide).
6.
[2 pts] Draw the repeat structures of cellulose and starch. Which one is
thermodynamically more stable? Why?
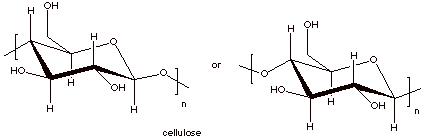
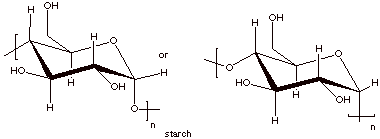
IV. Lipids
A.Triglycerides
1.
[3 pts] Draw a generalized structure
for a triglyceride which might be found in nature as an animal fat or vegetable
oil. Indicate as specifically as
you can the kinds of carboxylic acids (fatty acids) which are found, in
combined form, in triglycerides.
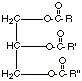
q
The fatty acids are
from about C12-C20 and are predominantly the even numbered ones.
2. [2 pts] Provide the full structures and common names of two saturated fatty acids which are found abundantly in triglycerides.
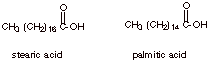
3.
[2 pts] Provide the full structures and common names of two unsaturated fatty
acids which are found abundantly in triglycerides.
q
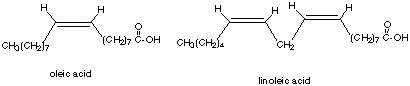 Note:
they should be cis.
Note:
they should be cis.
4.
[1 pts] Explain, with the assistance of an illustration, why triglycerides
which contain mostly saturated fatty acids are typically solids, whereas those
which contain mostly unsaturated fatty acids are liquids (oils).

q
The all anti
saturated alkane linkages fit together nicely and provide close approach and
favorable van der Waals attractive forces.
q
The cis double bonds
hinder close approach and diminish the van der Waals interactions (see dotted
lines for repulsions which impede close approach of the rest of the chain.
B. Soaps, Detergents and
Micelles
1.
[2 pts] Provide a structural illustration of a soap micelle, explaining what
factors stabilize the interior of the micelle and what factors stabilize the
exterior.
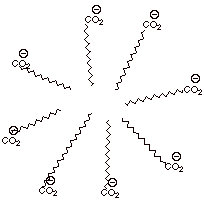
q
The van der Waals
attractions between the long hydrocarbon chains stabilize the interior.
q
The solvation of the
ionic carboxylate groups by the protic ends of water dipoles stabilizes the
periphery.
2.
[2 pts] Draw the structure of a
known detergent molecule as specifically as you can. Explain what advantage
such a detergent would have over soap.
![]()
q
Note: the Na ion need
not be included here.
q
The alkyl side chain
can have variable lengths, but about C8 is optimum.
q
The advantage is that
the sulfonate salts of calcium an d magnesium ions are soluble, whereas the
carboxylate salts of these ions are insoluble. Therefore, in hard water, soaps
precipitate out of solution and are ineffective, whereas detergents of this
kind are soluble and effective.
C. Terpenes and Steroids
1.
[3 pts]Terpenes are ultimately formed from acetyl coenzme A via isopentenyl
pyrophosphate, the structure of which is shown below. Sketch the mechanism of
the conversion of isopentenyl pyrophosphate to geranyl pyrophosphate.
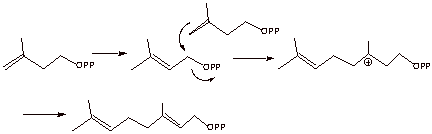
2.
[1 pt] What is the function of the pyrophosphate group in this reaction
sequence?
q
As a leaving group.
3.
[1 pt] In the first step of the mechanism, the double bond must migrate. In
terms of the nucleophilic substitution reaction which constructs the new
carbon-carbon bond, why is it vital that the double bond first migrate to the
new position?
q
This makes the pyrophospate
leaving group in an allylic position, so that it can undergo facile SN1
or SN2 substitution.
4.
[2 pts] In this same key step, isopentenyl pyrophosphate acts as the carbon
nucleophile. Give two reasons that it is especially suitable for reaction with
the developing carbocation.
q
The terminal carbon
of the double bond is an unsubstituted methylene group, which has minimal
steric hindrance toward the entering electrophile.
q
The carbocation which
will be formatted is tertiary, and thus relatively favorable.
5.
[1 pts] Starting with geranyl pyrophosphate, explain in broad general terms
(without a detailed mechanistic sketch) how triterpenes are formed.
q
Geranyl pyrophosphate
is first converted to farnesyl pyrophosphate by reaction with another molecule
of isopentenyl pyrophospate, as above.
q
Two molecules of
farnesyl pyrophosphate are coupled to give a triterpene.
V. Amino Acids, Peptides, and Proteins
A. Structures and Acidity/Basicity Considerations
1.
[1 pt]Write the structure of the most stable form of alanine (The less stable
structure is shown below).
![]()
![]()
2.
[1 pts] Draw the Fischer structure of L-alanine.
![]()
3.
[3 pts] In the inventory of amino acids shown below, indicate whether each is
acidic, basic, or neutral, and the approximate pKI of each.
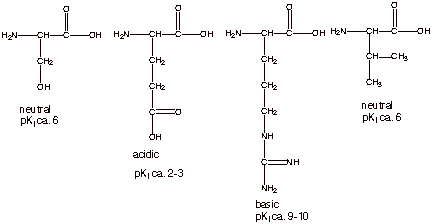
4.
[2 pts] The pKa of the conjugate acid of alanine is about 2. Compare
this to the pKa of a typical carboxylic acid and explain the
difference.
q
The pKI of
the alanine conjugate acid is about 2 (between 2-3 is acceptable), whereas that
for a typical carboxylic acid is about 5. So the alanine conjugate acid is much
more acidic.
q
The basis for this is
that the NH3+ substituent on the alanine conjugate
acid is a strong EWG, which highly stabilizes the anion (conjugate base) and
thus increases the acidity. (Or, the positive charge of the NH3+
substituent electrostatically attracts the negative carboxylate anion of
the conjugate base and strongly stabilizes it.)
B. Amide Structure and Bonding
1.
[3 pts] Draw two additional resonance structures for the amide function shown below and summarize as a
dotted line/partial charge structure. Explain why amide resonance is stronger
than ester resonance.
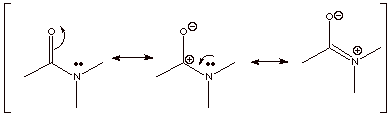
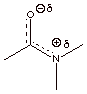
q
Note: A partial
positive charge may also be placed on the carbonyl carbon.
q
Amide resonance is
stronger than ester resonance because the resonance structure with positive
charge on nitrogen is a better structure (lower energy) than that with positive
charge on oxygen (electronegativities).
2.
[1 pts] Based upon your DL/PC structure from the preceding question, explain
why amide functions form much stronger hydrogen bonds than do esters or other
functionalities.
q
The oxygen is a
better H-bond acceptor because of the large partial negative charge.
q
The N-H bond is a
better H-bond donor because the nitrogen is positively charged.
3.
[1 pt] In a similar way, explain why rotation around the carbonyl-to-nitrogen
bond is very difficult.
q
The structure with
positive charge on nitrogen also has a double bond between the carbonyl carbon
and the nitrogen. Because this structure contributes so much to the resonance
hybride, there is a large amount of double bond (pi bond) character to this
bond. Consequently, rotation around this partial pi bond is difficult.
4.
[2 pts] Draw two different conformations (energy minima) for the amide function
shown below. Indicate which is more stable and why.
![]()
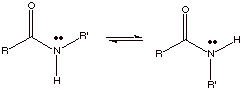
q
The first (trans) is
more stable, because the second (cis) has R and R’ groups that exert a
steric interaction between them
5.
[2 pts] For a general amide function, indicate via an illustration how many
atoms are required to be coplanar. Explain why, being as specific as you can.
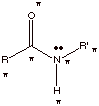
q
Six atoms (the
starred ones), including the carbonyl carbon and oxygen, the nitrogen and its
attached H and carbon and the carbon attached to the carbonyl group.
q
The carbonyl carbon
is sp2 hybridized so all three atoms attached to it are coplanar.
The amide nitrogen is also sp2 hybridized, so all three atoms
attached to it are coplanar. Because of amide resonance the trigonal plane of
the N and of the carbonyl carbon are required to be coincident. Therefore all
six atoms involved are coplanar.
C. Peptides: Nomenclature and Synthesis
1.[
2 pts] Provide the structures and
the names (using three letter designations of the amino acids) of two different
dipeptides which could be formed from one molecule of L-glycine and one of
L-alanine.
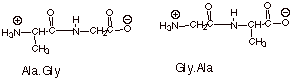
q
Note: the covalent
forms are equally acceptable.
2. [
5 pts] Sketch a selective
synthesis of the dipeptide gly.ala, starting with the respective amino acids.
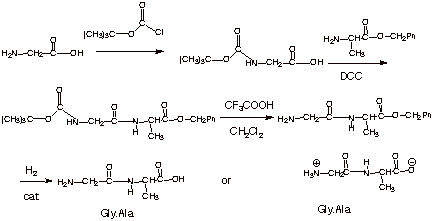
q
Abbreviations may be
used, e.g., Boc for the tert-butoxycarbonyl group and Bz for the benzyl ester.
q
HF or HBr may also be
used to cleave the benzyl ester.
3.[
2 pts] Write the mechanism for the
cleavage of the Boc protecting group to a carbamic acid by trifluoroacetic
acid. Why doesn’t this cleave the peptide functionality?
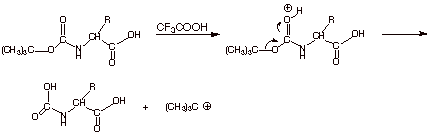
q
Doesn’t cleave
a peptide moiety because it is non-aqueous. Amide hydrolysis requires water.
D. Merrifield Solid Phase Peptide Synthesis
1.[
4 pts] Starting with the
Merrifield resin and the required protected amino acids, sketch a synthesis of
the same dipeptide, gly.ala. You may use an appropriate abbreviation for the
Merrifield resin.