CHAPTER 21: AMINES
DEFINITION:
Amines are organic derivatives of ammonia, in which one, two, or all three of
the hydrogens of ammonia are replaced by organic groups. Compounds RNH2
are called primary amines, R2NH secondary amines, and R3N
are tertiary amines.
q
Important
Note: The designation of amines as primary,
secondary, and tertiary is different
from the usage of these terms in connection with alcohols and alkyl halides. In
these latter two cases there is only one organic group (R), so that the terms
are used to designate the type of carbon to which the alcohol or halide
function is attached.
q
Consequently, tertiary
butylamine is a primary amine, but tertiary butyl alcohol is classed as a tertiary alcohol.
q
Similarly, dipropylamine
is a secondary amine, even though the R groups attached to nitrogen are
primary.
NOMENCLATURE: There
are two valid systems for naming amines. One system is used for naming relatively simple amines, i.e., molecules
in which there are no other functional groups than the amine function and the
compound is named as an amine, while the other is more flexible for naming
molecules in which there are functional groups other than amines or in which
there is considerable molecular complexity, so that it is convenient to use the
amino functional group as a substituent.
Simple
Amines. Simple amines are named according to the number of
carbons in the longest continuous chain (or ring) of carbon atoms present in any of the R groups attached
to nitrogen. Numbering, of course,
must begin with the carbon immediately attached to the amine function. After
identifying the main chain, the amine is named as a derivative of the alkane
(or cycloalkane) having the appropriate number of carbon atoms by deleting the
terminal –e of the alkane and replacing it with the suffix –amine. For example CH3NH2 , the
simplest amine, is named methanamine.
The common name for this very simple amine is methylamine (no separators between methyl and amine).
The secondary amine which has one methyl group and one ethyl group
attached to nitrogen is named N-methylethanamine (the two carbon chain is used
as the main chain in preference to the one carbon chain). Note the usage of the
letter N to designate that the methyl substitutent is attached to nitrogen. If
one R group is methyl, a second is ethyl, and a third is propyl, the amine
would be named N-ethyl-N-methylpropanamine. Note the alphabetic criterion for
arranging the methyl and ethyl sequence.
More Complex
Amines. The substituent name of the
–NH2 is amino.
The (CH3)2N- substituent is, e.g., N,N-dimethylamino. The
name of the compound
H2N-CH2CH2CH2OH
is therefore 3-amino1-butanol.
REACTIONS
OF AMINES
Basicity. You will
recall that the nitrogen atom of ammonia is sp3 hybridized and there is an unshared pair of electrons in the fourth tetrahedral orbital. This makes ammonia
a base and a nucleophile. Because
nitrogen is less electronegative than oxygen, ammonia is a much stronger
base than water and also a much better nucleophile. Amines, which are merely organic derivatives of
ammonia, are also tetrahedrally hybridized and are comparably basic and
nucleophilic to ammonia. You might recall that amines are completely
neutralized (protonated) by carboxylic acids.
The
basicity of amines is often discussed indirectly in terms of the acidity of their respective
conjugate acids. Recall that the
conjugate acid of a weak base (e.g. like water) is a strong acid (like
hydronium ion), while the conjugate acid of a strong base (like hydroxide ion)
is a weak acid (like water). The concept of pKa has already been developed as a measure of the
acidity of Bronsted acids, and we will also see that a corresponding concept, pKb can be used as a measure of the basicity of bases and
that these two quantities are very closely related. Consider the acid dissociation, in dilute aqueous
solution, of ammonia and a representative primary, secondary, and tertiary
amine:
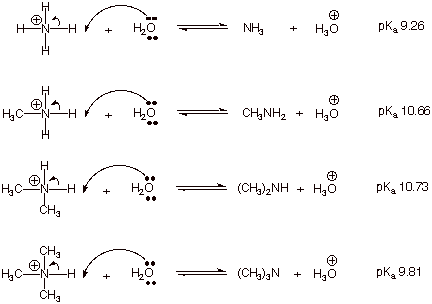
q
Note that the strongest
acid (least positive pKa) is ammonia. This means that ammonia is the
weakest base of the four bases.
q
We can easily understand
this because alkyl groups are electron donating (EDG), so they stabilize the positively charge
ammonium ions, i.e., the methyl ammonium ion is more stable than the parent
ammonium ion because the alkyl group stabilizes the positive charge on the
attached nitrogen atom.
q
Note also that the alkyl
stabilizing effect is purely inductive! [By looking at possible resonance structures, see if you can see why
there is no hyperconjugative resonance stabilization by the alkyl group.
q
Notice that the second
alkyl group, in the dimethylammonium ion, has only a very slight effect, while the third group (in the trimethylammonium ion)
causes an increase in acidity
(decrease in basicity) relative to the dimethylammonium ion. Of course, the
trimethylammonium ion is still less
acidic than ammonia.
q
All of the amines are
more basic than ammonia, but primary
and secondary amines are the most basic.
q
The effect of the third
alkyl group is another instance of steric inhibition of solvation. The presence of three alkyl groups sharply diminishes
the ability of the solvent to stabilize the corresponding ammonium ion, thus
causing a reversal in the tendency of the alkyl groups to decrease acidity and
increase basicity.
q
Please note the
relationship between pKb and pKa is pKa + pKb
= 14 in water.
q
The definition of pKb
is shown below:
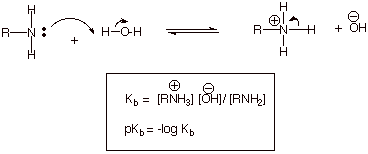
q
The pKb’s
of ammonia, methyl amine, dimethylamine, and trimethyl amine are therefore,
respectively, 4.74, 3.34, 3.27. and 4.19. Note that, in terms of pKb,
the strongest bases have the least positive values of pKb, just as
was the case for acidity in its relationship to pKa’s.
ACIDITY AS A
BASIS FOR SEPARATING AMINES FROM ORGANICS HAVING OTHER FUNCTIONALITIES. Amines
are the most basic class of organic
compounds. They are virtually the
only organic compounds which are substantially basic in aqueous solution and
which are completely protonated by dilute solutions of strong acids. Upon
protonation, of course, the form salts of the alkyl ammonium ions, which are
water soluble (if the R groups are
not too large). Consequently, amines can be separated from other classes of
organic compounds like halides, ethers, alcohols, and ketone (as well as
alkanes, alkenes and alkynes, of course), by a simple extraction technique.
q
The solution of the
mixture of organic compounds dissolved in an organic solvent such as ether is
treated with dilute aqueous acid (careful: exothermic).
q
The amine is protonated
and goes into the aqueous solution as an ammonium salt, while other
functionalities such as ketones remain in the organic phase.
q
The phases are separated
(separatory funnel), and the non-amine organic compounds are obtained from the
ether phase (drying and evaporation of the ether), while the amine is obtained
from the aqueous solution by adding more ether and making the aqueous solution
alkaline, which liberates the amine, this dissolving in the ether phase. After
drying and evaporation, the amine is obtained.
q
Note that this would of
course not work if the ketone or alcohol has only 1-4 carbons, because an
alcohol or ketone having such few carbons would have substantial water
solubility.
ACIDITY OF
AMINES. Note that
primary and secondary amines, like
ammonia have protic hydrogens and therefore possess a degree of acidity (unlike
tertiary amines, which have no acidic hydrogen). We have previously seen that
ammonia has a pKa value of about 38, and is a very weak acid.
Primary and secondary amines have pKa’s of very similar
magnitude. Consequently, such amines are much more basic (pKb about
4) than they are acidic (pKa 38), so that their aqueous solutions are rather strongly alkaline.
q
Since amines are only
very weakly acidic, their conjugate bases, RNH- or R2NH-
are very strong bases!! We have seen
that they are strong enough bases to be able to generate enolates of ketones
quantitatively.
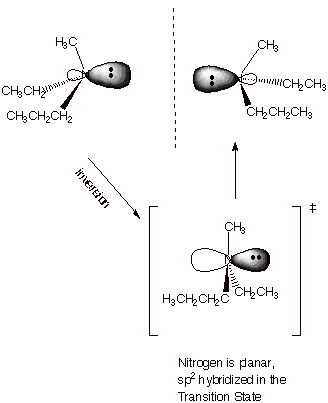
CHIRALITY OF
NITROGEN. It is
interesting to note that, since the nitrogen atom of amines is tetrahedral,
such a nitrogen can be a stereocenter if it has three different R groups
attached. By definition, the fourth group is an electron pair, so that all four
groups are different.
q
However, it is observed
that when chiral amines are generated, they very rapidly undergo an
umbrella-like inversion to generate the corresponding enantiomer, quickly racemizing
the amine. Certain amines, for which this inversion is especially difficult,
can be prepared and are relatively stable as a single enantiomer.
q
Please note, however,
that if a fourth different R groups is added in the context of a tetraalkyl
ammonium ion, this kind of inversion is prevented, and such quaternary ammonium
ions can be chiral and stable as a single enantiomer.
SYNTHESIS OF
ALIPHATIC AMINES (Aliphatic means the
groups attached to nitrogen are alkyl or cycloalkyl, but not aromatic as in the
case of aniline).
q
Since amines are fairly basic functional groups, it stands to reason that they are
also fairly nucleophilic.
q
Since amines are far more
basic than any oxygenated functional group such as an alcohol or an ether or
ketone, they are also expected to be, and are, much more nucleophilic than this oxygenated functionalities. Consequently,
they can be used effectively as nucleophiles in SN2 reactions with
alkyl halides. This also applies to ammonia, the inorganic parent of organic amines.
q
Using ammonia as a
nucleophile in a reaction with an appropriate (methyl, primary, or secondary)
alkyl halide in an SN2 reaction to prepare primary amines does work,
but it requires a huge excess of ammonia, because the product primary amine is also reactive toward the
alkyl halide. This would produce a
secondary amine, and then even further reaction with alkyl halide would give a
tertiary amine. Thus, a mixture of primary, secondary, and tertiary amines
would be generated unless ammonia is used in large excess.
q
When ammonia is present
in large excess (e.g., at least 10 fold) over the alkyl halide, the alkyl
halide has much more ammonia to react with than it does the amine.

q
It should be noticed
that the initially formed product is an alkylammonium cation, which can not act
as a nucleophile (no unshared electron pair), so it could not react, itself,
with alkyl bromide to give a dialkylamine.
q
However, in the presence
of ammonia, the proton transfer shown below, which produces the free alkyl
amine (which is a nucleophile and can react with alkyl bromide to give a secondary amine)
and the parent ammonium ion is quite rapid (remember: proton transfer from one
electronegative atom to another is very fast).
![]()
q
An alternative route for
forming amines---specifically and exclusively primary amines--- is to employ
another nitrogen nucleophile which is readily available, the azide anion. This reacts readily with an alkyl halide to give an
organic azide, which can be reduced with lithium aluminum hydride to the
primary amine. We will not look into the specific mechanism of this latter
reduction reaction.
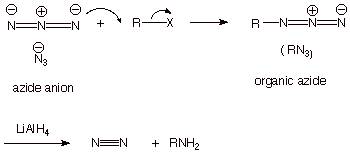
q
This strategy works
because the azide anion is a strong nucleophile, but the neutral organic azide
is a very weak nucleophile (recall that hydroxide anion is a strong
nucleophile, but its neutral conjugate acid, water, is a very weak
nucleophile). Therefore, the organic azide, once formed, is unable to react
with the alkyl halide. The result is that we do not have to use an excess of
the nucleophile to get exclusively
the primary amine.
q
We want to note that the
cyanide anion (which is a carbon type nucleophile which contains nitrogen) is a
strong nucleophile which can readily react with alkyl halides to produce
organic cyanides, which are called nitriles. These nitriles can also be reduced with lithium
aluminum hydride to the primary amine. In this case, the primary amine has one
additional carbon atom than is contained in the alkyl halide.
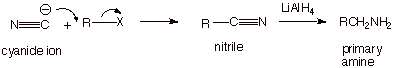
q
Amides can also be
reduced in the same way as nitriles.
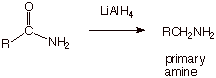
ANILINE
AND ARYLAMINES.
Synthesis.
q
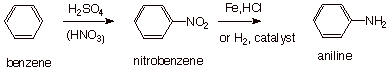
Many arylamines can be
synthesized by first installing a nitro function (another nitrogen-containing functionality which is
easily introduced onto an aromatic ring, as you know) and then reducing it to
the amino function.
q
Aniline (which is
essentially phenylamine) is the simplest aromatic amine. It can be synthesized
as shown below.
Basicity.
q
As an amine, aniline
(and its related arylamines) are basic.
q
However, it is much
less basic than typical alkyl amines.
Replacing an alkyl group by a phenyl or other aryl group greatly diminishes the
basicity of the amine function.
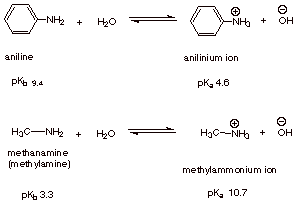
q
Note that the pKa
of the anilinium ion (the conjugate acid of aniline) is 4.6, whereas that of
methylamine is 10.7. This means, of course, that the anilinium ion is a one-millionfold stronger acid than the methylaminium ion.
Correspondingly, this means that aniline is a weaker base than methylamine, by
a factor of a million!
THE
THEORETICAL BASIS FOR THE DIMINISHED BASICITY OF ANILINE
q
Recall that
stabilization of the reactant side of the equation tends to diminish acidity
(because the hydronium ion is on the right hand side of the equation), while
stabilization of the product side tends to increase acidity.
q
Aniline is rather
strongly stabilized by resonance, whereas the anilinium ion is not. The
resonance structures for aniline are shown below, where it is shown that the
ring becomes electron rich, with partial negative charge (carbanion character)
at the ortho and para positions, while the nitrogen tends to become electron
deficient (partial positive charge).
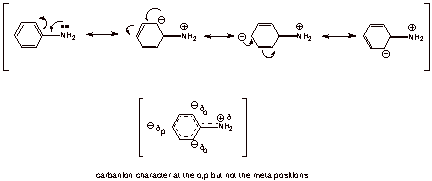
q
This resonance or
delocalization stabilization is possible because the unshared pair of electrons
on nitrogen are in conjugation with (able to directly overlap with) the 2p AO
on the directly attached ring carbon. These electrons are then delocalized
around the ring on to the positions indicated. See the indicated overlap in the
orbital picture shown below:
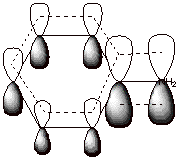
q
This conjugation is only
possible when the orbital external to the ring is in the benzylic-type position
(that is, on an atom directly attached to the ring).
q
So the reactant is
resonance stabilized in the case of aniline, but of course not in the case of
methylamine, which does not have a p type orbital available to overlap with. This
makes aniline much more stable thermodynamically than methylamine or any
alkylamine, and thus much less readily protonated (weaker base).
q
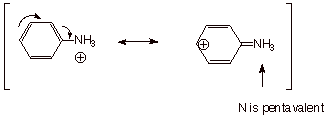
Finally, the
anilinium ion (the conjugate acid of aniline) lacks this conjugated system,
because the nitrogen atom is positively charged (highly electron deficient) and
thus it cannot contribute any electrons to the ring. Of course it could not
accept electrons from the ring because it doesn’t have any vacant
orbitals to use for such acceptance (this would violate the octet rule). Note
that the resonance structure on the right, below, is not a valid resonance
structure.
PYRIDINE. Pyridine is an aromatic amine, but in a very
different sense from aniline. Pyridine is essentially benzene with one of the
CH groups of benzene replace by a N atom.
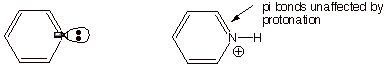
q
Note below that the
unshared electron pair in pyridine is in the trigonal plane, perpendicular to
the pi system consisting of overlapping pz AO’s, so the
unshared pair is not a part of the aromatic system, but is independent of it.
q
Pyridine, like aniline, is much less
basic than typical aliphatic amines,
but for a very different reason: the unshared pair is in an sp2 AO,
which as you recall is much lower in energy than the electron pair of aliphatic
amines, which is in an sp3 AO. Therefore, pyridine is less easily
protonated than typical aliphatic amines such as piperidine. The pKa
of pyridine is 5.25.
q
Interestingly, the
pyridinium ion (the conjugate acid) remains aromatic, because when the unshared
pair bond to a proton, the C-H bond is in the trigonal plane, and doesn’t
remove any electrons from the pi electron system, which remains a 6 pi electron system, like benzene.
Reactions of
Amines.
- The Hoffmann Elimination Reaction. Recall that alkyl halides (except fluorides) and
alcohols (in the presence of acid) can undergo elimination reactions to
give alkenes. In both of these systems, good leaving groups are present,
thus permitting an E2 elimination (or in some cases an E1 elimination). In
the case of halides, the chloride, bromide, and iodide ions are good
leaving groups. Recall that good leaving groups are weak bases. In the case of alcohols, the hydroxide
ion, being a strong base, is a poor leaving group, but in acidic solution,
when protonated, a good leaving group is generated (water). What about
amines. If we should want to perform an elimination reaction on an amine
to convert this functionality to an alkene function, could we do it? And
how could we do it?
q
First, we note that the
amide ion (NH2--) is even more strongly basic than a
hydroxide anion, so it would be an atrocious leaving group.
q
What about using acid,
as in the case of alcohols, to generate a better leaving group? In the simplest
case, this would be ammonia (NH3), which is not too strong a base (albeit
more basic than water or a halide ion).
q
In acidic solution, we
would not have a very good base to abstract the beta proton, we would have to
settle for water as the base. The question is, is ammonia a good enough leaving
group to effectively leave when the weak base water is the best base available.
It is not! The difference between
the eliminations of alcohols and amines in acidic solution is the poorer
leaving group ability of ammonia than that of water (remember, ammonia is a stronger base; therefore a
poorer leaving group.)
q
What is the solution to
this apparent dilemma? Essentially, convert the ammonium ion function to a
functional group which will allow the use of a strong base, like hydroxide
anion. Since the amide ion is such a terrible leaving group, it would still
have to be converted to the ammonium form, so that the leaving group could
be a neutral amine. This can
only be done if all of the acidic protons of the ammonium ion are removed and
replace by alkyl groups, specifically methyl groups.
q
Since amines are pretty
decent nucleophiles, as well as bases, they can react with alkyl halides in an
SN2 displacement, as shown below.
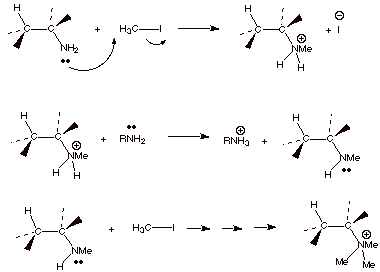
q
The primary amine is
first converted to a secondary amine function, the secondary amine to a
tertiary amine, and finally this reacts with a third molecule of methyl iodide
to give the quaternary ammonium salt. At this point, there are no more acidic
protons, so base can be employed in an E2 reaction.
q
This is usually done by
first reacting the quaternary ammonium iodide, which is initially formed to a
quaternary ammonium hydroxide, by treatment with silver oxide (giving insoluble
silver iodide). At this point we have a good base and a reasonable leaving
group. Heating this ionic compound up to arount eighty degrees usually succeeds
in effecting elimination of trimethylamine.
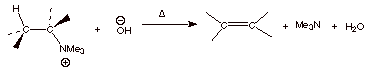
Transition
State for the Hoffmann Elimination Reaction.
q
Like all E2 reactions,
this reaction is concerted.
q
We would like, then, to
develop a transition state model for the reaction, so we can rationalize and/or
make predictions of such things as selectivity (especially regioselectivity).
q
We do this in the usual
way, using canonical structures for the reactant and the product, but also for
a non-reactant and non-product-like structure (an “X” structure).
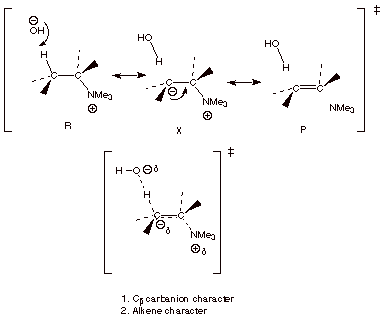
CARBANION AND ALKENE CHARACTER IN THE TS’S FOR ELIMINATION REACTIONS.
q
Note that when we
derived the TS model for the elimination of HX from an organic halide by a base
such as hydroxide ion, for simplicity we used only the reactant and product-like structures, so we only were
able to see the alkene character of the TS. Fortunately, it is the alkene
character which is dominant in the
eliminations of alkyl halides.
q
However, some
carbanion character is also present in that type of elimination, and in all
such eliminations. The importance of including the more complete treatment
which reveals the carbanion character in the present instance (eliminations
where the leaving group is an amine) is that it is now the carbanion character
which is dominant over alkene character, resulting in a sharp change in the
regiochemical selectivity.
q
Recall that the
elimination of alkyl halides tended to favor the more highly substituted
alkene which is the more stable
alkene. We called that Saytzeff
regiochemistry.
q
In the Hoffman
elimination reactions of alkylammonium hydroxides, the less stable alkene is favored, fundamentally as a result of more
favorable carbanion character. This kind of regiochemistry is called Hoffmann
regiochemistry.
q
For example, in the
elimination of the quaternary ammonium salt shown below, 1-butene is very
strongly favored over the 2-butenes, even though the alkene character in the TS
would tend to favor the latter.
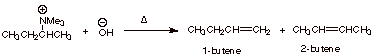
q
The TS models for the
two competing TS’s are shown below:
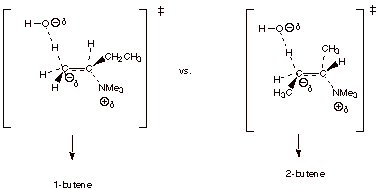
q
The TS leading to
1-butene is favored, because it has primary Cb carbanion character, while that leading to 2-butene has secondary
carbanion character.
q
Carbanions are
destabilized by alkyl groups (remember that alkyl groups are electron donating
groups; they stabilize positive charge, but destabilize negative charge. The
order of carbanion stability is:
methyl more stable than primary than secondary than tertiary.
q
Note that the TS
leading to 1-butene has primary carbanion character, while that leading to
either of the 2-butenes has secondary carbanion character.
q
The ratio of 1- to
2-butenes is approximately 90:10.
q
Finally, we raise
the question of why the drastic change in the relative amounts of carbanion and
alkene character in the two types of elimination (alkyl halides and
alkylammonium salts). Note that of the three canonical structures for the TS,
the one which gives rise to alkene character is the last one (in our drawing
above). The structure will be of lower energy and contribute more when the
leaving group is of lower energy (in this structure the leaving group has
left.). That is, the better the leaving group the more alkene character
there is in the TS. Since chloride (or bromide or iodide)
ions are better leaving groups than trimethylamine, the alkyl halide
eliminations have much more alkene character than do the alkylammonium ion
eliminations.
q
Interesting that
fluoride ion, the worst of the halogen leaving groups, tends to give
regiochemical preferences which are more like those of the alkylammonium ions,
i.e., favoring the less substituted, less stable, alkene.
DIAZONIUM
IONS AND THEIR REACTIONS.
All
primary amines are readily converted by nitrous acid to diazonium salts. In the
case of aliphatic R groups, the diazonium ions are extremely unstable, rapidly
decomposing to give carbocations which undergo reaction with whatever
nucleophiles may be present (such as water). The reason this especially high
level of reactivity is that dinitrogen, being thermodynamically highly stable,
is an outstanding leaving group. [Recall, again, good leaving groups are weak
bases, and nitrogen is a really poor base]
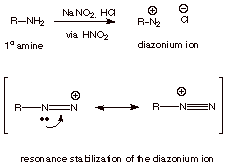
q
Note that the positive
charge in the diazonium ion is delocalized, i.e., it is shared by both nitrogens. Positive charge on nitrogen is inherently not very
favorable (electronegative atom), but resonance stabilization makes this ion
stable enough to form.
q
However, when the R
group is alkyl, these diazonium ions readily decompose via an SN1
mechanism, with dinitrogen as the leaving group.
q
Note that because
nitrogen is such a thermodynamically stable molecule, it is perhaps the very
best leaving group of all. Even when
these diazonium ions are formed at ice bath temperatures, they lose nitrogen
extremely quickly, forming a carbocation, which then reacts with available
nucleophiles (e.g., solvent or chloride ion).
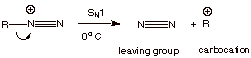
q
However, when R is an
aryl group, such as phenyl, the diazonium ion is moderately stable at about
zero degrees centigrade, but when
warmed up to room temperature it rapidly decomposes en route to room
temperature. This permits the use of the aryldiazonium ions in reactions with
substances supplied after the diazonium ion is generated.
q
Aryldiazonium ions are
more stable than alkyldiazonium ions because the Ar-N bond is partially double,
as shown in the resonance structure below, which is an additional small
contributor over the main two resonance structures written previously. In other
words, the pi system of the N-N pi bond overlaps with the pi system of the
benzene ring, providing delocalization of the positive charge onto the ortho
and para positions of the benzene ring.
![]()
Azo Compounds
q
Aryldiazonium ions are
electron deficient and therefore are electrophilic. However, they are relatively mild (not highly reactive, but very selective) electrophiles, because of their resonance
stabilization.
q
One of their main uses
in in electrophilic aromatic substitution reactions. As very mild and selective
electrophiles, they do not react with benzene or toluene or even anisole (methoxybenzene—normally
considered a highly reactive aromatic). They do, however, reactive with
aromatics which have the powerfully electron donating amine function. In
particular, N,N-dimethylaniline reacts readily with aryl diazonium ions as shown
below:
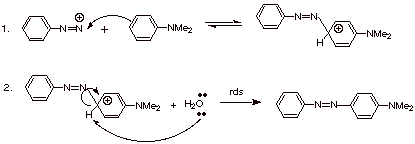
q
The final product of
this reaction is generically called an azo comound.
q
Azo compounds are highly
colored compounds and are frequently used as dyes for textiles.
The Sandmeyer Reaction
Another common use for aryldiazonium ions is in the
transformation of the amino group of aniline or a derivative of aniline to
other functionality such as a halide or a nitrile function. This involves the
addition of an appropriate salt containing the desired nucleophile to the cold,
aqueous solution containing the diazonium ion and the allowing the temperature
to ascend to room temperature. In this way, the diazonium ion decomposes to the
aryl carbocation, which then reacts with the appropriate nucleophile.
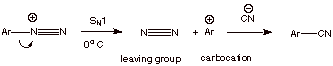
q
In this way, the amino
group can be converted to a chloro, bromo, iodo, or nitrile function (or even reduced to hydrogen by using an
appropriate reducing agent).
q
You may recall that aryl
halides do not under either SN1 or SN2 substitution
because the aryl halogen bond has double bond character and is too strong to
easily break. However, when the potent leaving group is dinitrogen, even
aryl systems can undergo an SN1 substitution reaction.