ELECTROCYCLIC REACTIONS
Electrocyclic Reactions
Pericyclic reactions are reactions in which a cyclic, conjugated system of electrons is created in the transition state. The Diels-Alder reaction is a common example. Electrocyclic reactions are a sub-type of pericyclic reaction which is unimolecular and in which the termini of a conjugated system become sigma bonded to each other to form a shortened pi system. The reverse of this, the cleavage of a sigma bond to generate a longer conjugated system, is sometimes called a retroelectrocyclic reaction. As an example of the latter, cyclobutene is cleaved thermally to yield 1,3-butadiene, relieving the extensive strain in the cyclobutene system and gaining the resonance stabilization of the conjugated diene system (Scheme 1). The alkene pi bond of cyclobutene is extended to give the conjugated system of butadiene. The especially interesting thing about this reaction is its stereochemistry, as revealed, for example, in the cleavage of cis- and trans-3,4-dimethylcyclobutadiene. The reaction is highly stereospecific, with the trans isomer of the reactant yielding the E,E isomer of the diene and the cis isomer of the reactant giving the Z,E isomer of the diene. Note that in the IUPAC nomenclature, E (entgegen) means trans and Z (zusammen) means cis. The observation of stereospecificity means that in both transition states (for the two isomers) a single rotatory pathway is followed, yielding different isomers of the product. In particular the groups attached to C3 and C4 must rotate in the same (both clockwise or both counterclockwise) direction. The term which is used in this connection is “conrotation”.
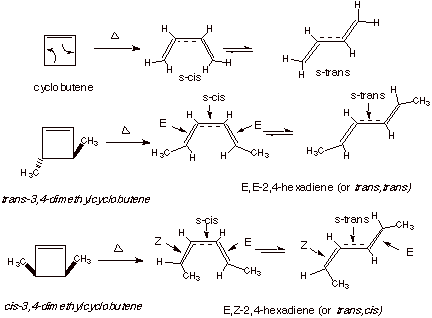
Scheme 1. The conrotatory electrocyclic reactions of cyclobutenes.
In cis-3,4-dimethylcyclobutene, e.g., the two methyl groups both are shown as rotating clockwise, as shown in Scheme 1 and also in Scheme 2, below. This yields the cis,trans (E,Z) isomer of the diene. On the other hand, in the trans isomer of 3,4-dimethylcyclobutene, if clockwise rotation is followed, the trans,trans (E,E)isomer of the diene is generated.
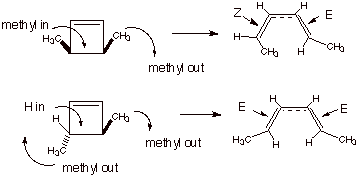
Scheme 2. Conrotation of C3 and C4 results in the formation of the E,E diene from the trans cyclobutene and the E,Z diene from the cis cyclobutene.
Note what is not observed, viz., disrotation, in which the groups attached to C3 and C4 rotate in opposite directions (one clockwise and one counterclockwise; shown in Scheme 3). In such a case, the cis cyclobutene isomer would give rise to E,E-2,4-hexadiene and the trans cyclobutene isomer would yield E,Z-2,4-hexadiene, i.e., the opposite results from what are actually observed. So the reactions, as experimentally observed, are not disrotatory, nor are they a mixture of disrotatory and conrotatory paths, but they occur exclusively by a stereospecific conrotatory path. It will be of interest to examine the reasons for this preference. Incidentally, electrocyclic reactions in larger (or smaller) rings always have a strong rotatory preference, but not always for conrotation. That is, some strongly prefer disrotation, as we shall see.
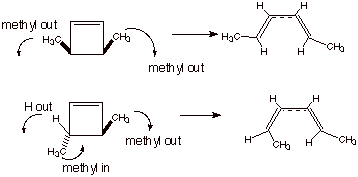
Scheme 3. The disrotatory cleavage of cyclobutenes which is not observed.
What is the basis for the strong preference for conrotation over disrotation? To see this, we can again use the theory of transition state aromaticity/antiaromaticity as applied to both of the competing transition states (Scheme 4).
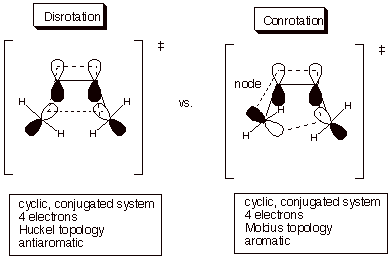
Scheme 4. The topologies associated with the conrrotatory and disrotatory reaction modes.
As you will recall, the normal (Huckel) cyclobutadiene MO display is predicted by the circle mnemonic in which a polygon of appropriate size is inscribed, with one apex down, inside a circle of radius 2b. In the Mobius topology, in which every MO has at least 1 nodal plane, the appropriate circle mnemonic is that in which the polygon is inscribed with one side down, as illustrated below (Scheme 5). Therefore, Mobius cyclobutadiene has two bonding MO’s and can support four electrons.
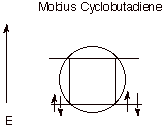
Scheme 5. The circle mnemonic for Mobius cyclobutadiene
In the case of the electrocyclic reaction of 1,3-cyclohexadiene to 1,3,5-hextratriene the converse result is observed, viz., disrotation (Scheme 6). This is as expected because the disrotatory mode maintains a normal Huckel-type topology, and with six electrons, this is aromatic. It is also pertinent to note that the actual reaction occurs in the forward electrocyclic sense, from hexatriene to cyclohexadiene, since the latter has little strain, and the conversion of a pi bond to a sigma bond is favorable in terms of bond strength.

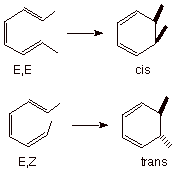
Scheme 6. The disrotatory electrocyclic reaction of hexatrienes to give 1,3-hexadienes.
The stereoelectronic relationships can perhaps be best seen by starting with the cyclic form (Scheme 7):
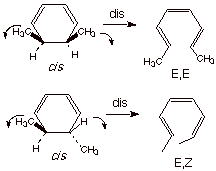
Scheme 7. Illustrating
how the disrotatory reaction mode converts E,E-2,4,6-hexatriene to cis
-5,6-dimethyl-1,3-cyclohexadiene and the E,Z triene to trans-5,6-dimethyl-1,3-cyclohexadiene
(looking at the reaction in the reverse sense)
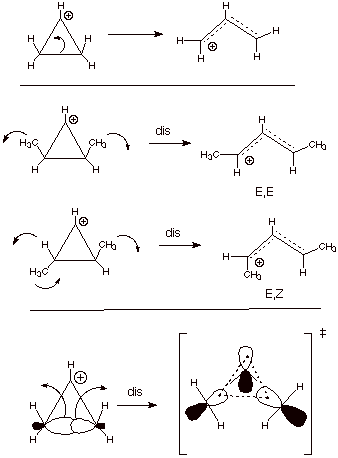
Retroelectrocyclic Reaction of the Cyclopropyl Carbocation. An excellent example of a retroelectrocyclic reaction which occurs via a stereospecific disrotatory mode is the cleavage of the cyclopropyl cation to the allyl cation (Scheme 8). Again, much strain is relieved and the large resonance stabilization of the allyl cation is gained. The stereochemistry is disrotatory because the cyclic, conjugated system present in the TS has two electrons and is aromatic in the Huckel topology.