Conjugated Dienes
A.Definition
q
A conjugated system
refers to a system of pi electrons
which consists of three or more 2pz AO’s on directly connected
atoms. The resulting pi MO’s are then delocalized over three or more
atoms.
q
A conjugated diene is a
molecule which contains two alkene
linkages which are in conjugation,
giving a four atom (also 4 AO) delocalized system.
q
Simple example of a
conjugated system is 1,3-butadiene, in which the two pi bonds are directly
connected so as to allow continuous overlap over the entire system of four
carbon atoms.
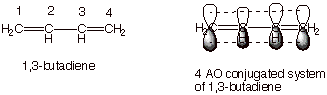
q
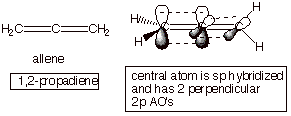
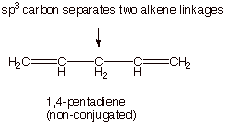
Dienes which are not
conjugated dienes are exemplified by 1,2-propadiene (allene) and
1,5-pentadiene. In the latter, the two alkene linkages are separated by a
saturated carbon atom, so that the pi overlap is not continuous. In the former,
the two pi bonds are perpendicular and do not interact or delocalize. But
1,3-pentadiene is a conjugate diene.
Nomenclature. The suffix “diene” is used to describe a
molecule having two “ene” linkages. A single number is used to designate the starting position
of each double bond, e.g. as shown below. Note that when naming a substance as
a diene, the numbering system of the parent chain begins at the end of the
chain closes to one of the double bonds.
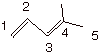
4-methyl-1,3-pentadiene
Conformations of
Conjugated Dienes.
q
In order to have a
conjugated system of AO’s, in which all of the 2p AO’s are parallel
(e.g., all 2pz), all 4 of the carbons of the diene system must be
coplanar, as we will see further in a moment.
q
This means that there
are only two ground state conformations of such a conjugated diene. One is
called s-trans and the other s-cis.
q
The cis or trans designation is based upon the
relationship around the central, formally single C-C bond.
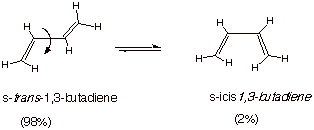
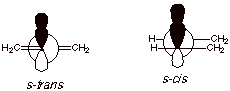
q
The s-trans conformation is more thermodynamically stable because
the s-cis conformation has a
steric repulsion between the two inside hydrogens.
q
However, both
conformations are found at least to some extent. We will see that although the s-trans conformation is preponderant, the s-cis conformation is extremely important in some reactions.
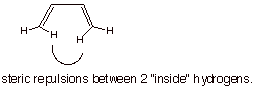
q
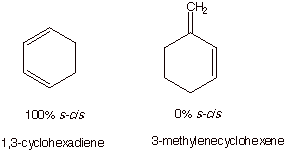
It should also be noted
that in some cyclic systems, this inside repulsion is not present, and the
molecule is rigidly compelled to have the s-cis conformation (no other stable conformation exists). In other
cyclic cases, the s-trans
conformation may be the only one available; i.e., there is 0% s-cis.
3. [2 pts] Using the above drawings, explain why these planar conformations are strongly favored over other, nonplanar, conformations. Show any relevant overlaps by means of orbital depictions.
Only in the planar conformations are the 2p AO’s at C2 and C3 parallel, so as to give maximum overlap and resonance stabilization. (See drawing above for orbital overlaps)
4.
[1 pts]Name one type of effect would normally tend to disfavor the planar
forms. Torsional effects.
5.
[1 pts] Which one of the two planar forms is the more stable? Explain, using a
structural illustration. The s-trans-conformation is the more stable one
because of the inside H/H steric repulsion in the s-cis conformation.
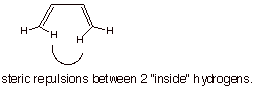
q
Note that one or both of
the ene linkages may have the capability of existing as either the cis or trans isomer. In such a case, the locant must
be preceded by a cis or trans or E/Z specification.
q
When a terminal carbon
of the conjugated system has a cis or Z substituent (anything other than
H), the s-cis conformation is
essentially unpopulated, because it is too high in energy.
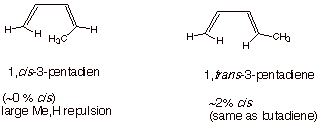
q
In both cases shown
above, the s-trans conformation is
predominant, but in the first case it is the exclusive conformer.
Resonance Stabilization of Conjugated Dienes
q
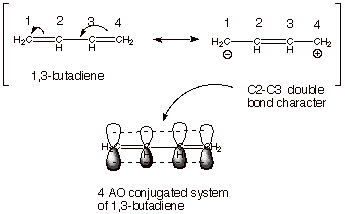
Since the four electrons
of the two double bonds are delocalized over a four atom conjugated system, we
would expect that this pi system is more stable than the sum of two alkene
double bonds. This is the case, although the stabilization is relatively small
(perhaps less than 8 kcal/mol) because the second resonance structure is
relatively higher in energy than the first one (one less bond; charge separation).
q
Nevertheless, this
resonance makes it relatively difficult to rotate around the central (C2-C3)
double bond, because at the 90o dihedral angle shown below, all
traces of resonance stabilization are lost, and the diene linkages are
independent (not conjugated). Rotation occurs, but requires about 8 kcal/mol, a
larger energy than required for rotation around typical C-C single bonds.
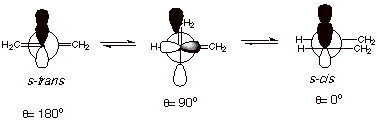
Conjugate Addition to Dienes. We are
familiar with the circumstance that H-X adds to alkenes to give alkyl halides.
It would, of course, not be surprising if a similar reaction occurs with
conjugated dienes. The mechanism of this electrophilic addition reaction is
shown below:
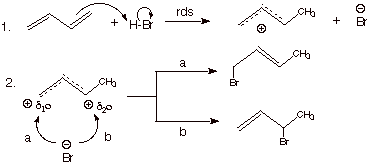
q
Interestingly, two
products are formed, in one of which HBr has in effect added to the 1 and 4
positions (the terminal positions) of the diene, with both of the original
double bonds being broken and replaced by one which has shifted to the central
carbons of the original diene. This is the first product shown above: trans-4-bromo-2-butene (also the cis isomer is formed in small amounts).
q
In effect the proton has
added to C1 and the bromide ion to the C4 position. This kind of product is
said to derive from conjugate addition to the diene.
q
In the other product
(3-bromo-1-butene), a proton has added to C1 and a bromide ion to C2. This is a
more or less normal 1,2 addition to an alkene double bond. The other double
bond remains unaffected.
q
The formation of two products is the result of the circumstance that the
intermediate carbocation (generically, an allylic type carbocation) is a
conjugated system in which the electrons are delocalized over 3 atoms, and the
positive charge over the two terminal atoms of the allylic conjugated system.
q
The nucleophile can
react at either site of positive charge, giving one or the other of the two
products.
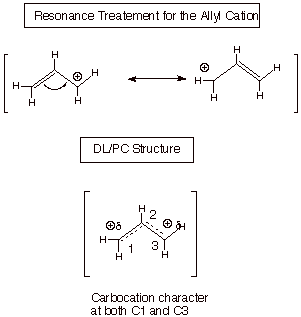
Allylic Resonance
Stabilization. Allylic resonance
stabilization is large, because in the parent allyl cation there are two equivalent resonance
structures. In other derivatives of the allyl carbocation, the structures may
not be exactly equal in energy but are close enough to give similarly large
resonance stabilizations. In the parent allyl cation, there is a half unit of
positive charge on C1 and also the same amount on C3, but none on C2. We can say
that there is extensive carbocation character at both of these terminal
carbons, so a nucleophile can react equally readily at either position (but not
at C2).
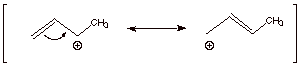
Unsymmetrical Allylic Carbocations
q
In the case of an
unsymmetrical allylic carbocation, the two resonance structures are not equal
in energy, and the real structure, while intermediate, will more closely
resemble the structure of lower energy.
q
In the addition to
1,3-butadiene, such an unsymmetrical allylic cation is formed.
Resonance Theoretical Treatment
Dotted Line/Partial Charge Structure
![]()
q
The first structure
drawn above is of lower energy than the second canonical structure, because the
first is a secondary carbocation canonical structure, while the second is a
primary carbocation canonical structure.
q
The real structure
(resonance hybride) more closely resembles the lower energy structure, which
has a secondary carbocation center.
q
Therefore, the resonance
hybrid has more carbocation character at the secondary than at the primary
carbon.
q
The bromide ion
therefore reacts preferentially (more rapidly) at the site of higher positive charge density. That
is, at the secondary carbon, to give:
![]()
q
However, the rates of
reaction at the two sites are not too different, so that the second product is
also formed in competition with the above product, by reaction at the primary
carbon. This product is the conjugate addition product.
![]()
q
This is kinetic
control (or, rate control). The two
products are formed in 80% and #0% amounts when the reaction is carried out at
low temperature. Under these conditions, the products, once formed, do not
equilibrate with each other, so that the observed product mixture represents
the ratio in which they were originally formed.
Addition of HBr to 1,3-Butadiene at Room Temperature: Thermodynamic Control
q
When the very same
reaction is carried out at room temperature, the same products are formed, but
the product ratio is significantly changed. Under these conditions, the
conjugate addition product (shown below) is the major product (about 80%) and
the 1,2-addition product is the minor product (about 20%).
![]()
q
It is not because the
reaction of bromide ion with the allylic carbocation has suddenly changed its
preference for reacting at the secondary site, but because at room temperature
these two products are equilibrating to give the thermodynamically more stable
product in excess.
q
The reason the conjugate
(1,4) addition product is thermodynamically more stable is that it has a
disubstituted alkene moiety, while the 1,2 addition product is only
monosubstituted.
q
The mechanism by which
the two bromides equilibrate is given below and simply involves the reformation
of the allylic carbocation/bromide ion pair.
.
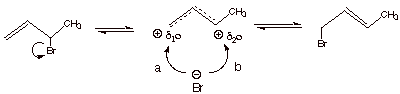
Thermodynamic vs. Kinetic Control: Testing
Suppose
two products, A and B, are formed in a certain reaction and product A is the
major product. You want to know whether this product mixture is kinetically
controlled or thermodynamically controlled. How could you test whether the
product mixture is kinetically controlled (therefore requiring an analysis in terms of Competing Transition
States to explain the preference for
one product or the other) or thermodynamically controlled (thereby requiring an
analysis of the relative stabilities of the two products).
q
We could, e.g., obtain
pure samples of A and B.
q
Subject each
individually pure isomer to the exact reaction conditions, including
temperature, solvent, reaction time, etc.
q
If A remains pure A,
without isomerizing, and B remains pure B, after this reaction time, the
product mixture was kinetically controlled. Essentially, this experiment shows
that both products are stable under the reaction conditions, once formed, and
do not rearrange into the other.
q
On the ther hand, if
pure A and pure B both rearrange to
the same mixture of A and B as obtained in the original reaction, the product
mixture is thermodynamically controlled.
Diels-Alder Cycloaddition
One of the most useful reactions in synthetic organic
chemistry is the Diels-Alder reaction, which consists of the reaction of a
conjugated diene (specifically in its s-cis conformation) with an alkene (preferably an electron
deficient alkene).
q
The Diels-Alder reaction
is especially important in organic synthesis for two main reasons. First, it
accomplishes the formation of two carbon carbon bonds. Secondly, it enables one to construct a cyclic, six-membered ring system. Both of these aspects
allow one to accomplish a dramatic increase in molecular complexity for the
purpose of synthesizing complex molecules.
q
Consider the prototype
(simplest) Diels-Alder cycloaddition reaction between 1,3-butadiene and ethane,
shown below.
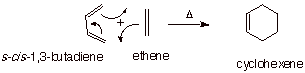
q
This reaction involves
breaking three pi bonds and forming one new pi bond plus two new sigma C-C
bonds. There is a net conversion of 2 pi bonds to two sigma bonds, so this is
highly favorable.
q
Recall that converting
two molecules into one is entropically unfavorable, but the large enthalpic
preference for sigma bonds is dominant.
q
In the context of a
Diels-Alder reaction, the alkene is referred to as the dienophile.
q
The reaction mechanism
is a concerted one, with all bonds being made and broken in a concerted way.
q
The TS model for this
reaction is developed below, using resonance theory, and R and P-like
structures.
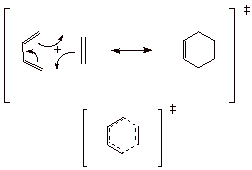
q
Characterization: The TS
has a cyclic 6e conjugated system. Four pi electrons come from the conjugated
diene system and two from the alkene pi bond.
q
Since 6e cyclic
conjugated systems are especially favorable (Huckel Rule), this is a relatively
favorable TS. [Not that is is an energy minimum, but the energy maximum is
lowered considerably by the stability of the pi electron system.
q
Such transition states
can be called “aromatic TS’s”.
q
Note that, since ethene
must bond simultaneously to Carbons 1 and 4 of the diene, the s-cis conformation of the diene is absolutely
required. The terminal atoms of
the s-trans conformer are much two
far apart to be bridged by a single ethene unit. Although the equilibrium
mixture contains only abut 2% of the cis conformer, the equilibrium is re-established rapidly, so that the
supply of the needed conformer is continuously and rapidly being restored.
q
Any diene which cannot
adopt the s-cis conformation for
some reason (as we have previously noted), cannot undergo the Diels-Alder
reaction. Dienes which exist only in the cis conformation are especially apt at undergoing the
Diels-Alder reaction.
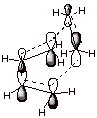
q
A more representative AO
model of the cyclic TS is given below:
Hypothetical Reactions
Which Would Have to Occur via an Antiaromatic TS.
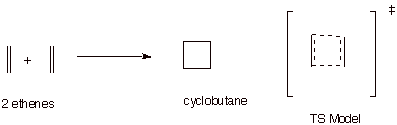
q
The reaction between two
ethene units to give a cyclobutane is thermodynamically more difficult than the Diels-Alder reaction because
of the ring strain present in the four-membered ring.
q
However, it is also
much, much less favorable kinetically because the TS is a cyclic system of 4
electrons, which is especially unfavorable. Such TS’s can be called antiaromatic
TS’s.
q
Although examples of
such reactions are known, they typically require powerfully forcing conditions
and, importantly, when they are
forced to occur they do not occur via a concerted pathway, so that they avoid
the unstable 4e system. [One bond is formed at a time, in a radical mechanism]
Diels-Alder Reactions with
Electron Deficient Dienophiles.
For reasons which we will not delve into, ethene itself and other simple alkenes are not the best dienophiles for Diels-Alder reactions. However, alkenes which have electron withdrawing substituents are much better. They react under much milder conditions ( sometimes at RT) and in better yield. Ester functions are among those which strongly accelerate the Diels-Alder reaction.
q
The equation below
illustrates the reactions of a pair cis/trans isomers containing two ester functions in the
dienophile with an especially reactive cyclic diene, which exists exclusively
in the s-cis conformation. The trans isomer of the dienophile is called dimethyl fumarate;
the cis isomer is dimethyl
maleate.
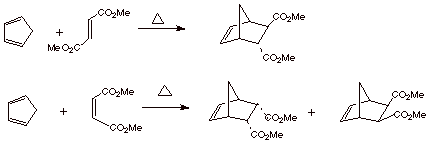
q
We especially want to
note the reaction stereochemistry. The
cis dienophile reacts to give two
products, both of which have the ester functions cis to one another, i.e., the stereochemistry present in
the original dienophile (cis) is
completely retained in the products. In the same way, the trans dienophile isomer gives a single product, which has
the ester functions trans to one
another. In both instances, the stereochemical relationships of the esters to
one another in the reactant are 100% retained in the product.
q
This signifies that the
addition to the dienophile (an alkene) has occurred with syn stereospecificity. Recall that syn stereospecific reactions are typically associated with
concerted mechanisms. The observed reaction stereochemistry is fully consistent
with the postulated concerted mechanism. Similar results have been observed for
a wide variety of Diels-Alder reactions, which are virtually always syn stereospecific.
q
If the reaction had been
stepwise, forming an intermediate diradical or other intermediate, it would be
expected that both dienophile isomers would have formed a mixture of all of the
three products.
q
In the reaction of
dimethyl maleate with 1,3-cyclopentadiene, the first isomer is strongly
predominant. The ester functions in this predominant isomer are described as
having the endo configuration,
which the ester functions in the minor isomer are described as having the exo
configuration. These two isomers are
more specifically diastereoisomers.
q
Typically,
Diels-Alder reactions have a preference for placing substituents in the endo
configuration. [We will not pursue the subject of why this is, at present]