Amino Acids, Peptides, and Proteins
Structure and Importance
Amino
acids are, in general, bifunctional molecules which contain both the amino (
-NR2) and carboxyl (-CO2H) functions, albeit not directly
connected to each other (as in an amide function). Amino acids are especially
important in nature, since they form the basis for peptides and enzymes.
Both of these latter classes of biochemically important molecules are
fundamentally copolymers of
various amino acids (about 20), in effect formed by condensation
copolymerization.
q
The amino acids which
are found in peptides and enzymes are exclusively a-amino acids, i.e., carboxylic acids in which the amino group is
attached to the alpha or C2 carbon, as shown for glycine (Gly) and alanine
(Ala).
q
A reasonably generalized
structure is given alongside the structures of glycine and alanine, indicating
that various organic substituents (in addition to hydrogen, as in glycine) are
present, also at the a position.
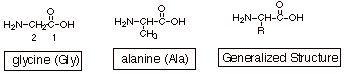
Fischer
Projection Structures of Amino Acids.
You may recall that Fischer structures were extremely
important in carbohydrate chemistry. They are perhaps less so in amino acid and
peptide chemistry, but are convenient for representing and discussing the configuration at the a carbon, which is a stereocenter for all naturally occurring amino
acids except glycine. You may recall the conventions for
writing Fischer structures include especially that horizontal bonds are
considered to be projecting toward the observer and vertial bonds away from the
observer.
q
Where Fischer structures
of amino acids are concerned, the carboxyl group is by convention written at
the top, in analogy to the aldehyde group of carbohydrates, and the R group is
written at the bottom, by analogy to the CH2OH group of a sugar.
q
The amino group is then
considered as analogous to the OH group of a sugar. When the amino group
projects to the left, the stereoisomer is considered to be the L enantiomer.
When it projects to the right, it is considered t be the D stereoisomer.
q
In nature, only the L
enantiomers of the amino acids are found.
q
Although the R,S
nomenclature system could also be used, the D,L system is mostly used for amino
acids, as it is for sugars. Incidentally, the naturally occurring L
enantiomer of alanine is the S enantiomer, according to the R,S system. You could verify this with molecular
models.
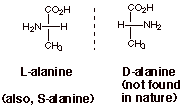
Four Sub-Classes of Amino Acids
q
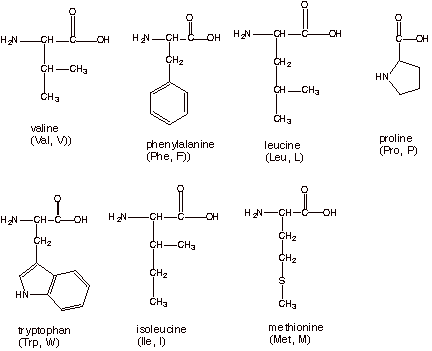
Most amino acids have neutral side chains (R groups). Many of these have R groups
which are nonpolar. Amino acids
which have nonpolar side chains are listed below (except for glycine and
alanine, which have already been illustrated.)
q
We will note,
momentarily, that even in the case where the R group is neither acidic nor
basic, the resulting amino acids are still slightly acidic, even though the
acidity of the carboxyl unit is partially neutralized by the basicity of the
amino unit.
q
Four of the naturally
occurring amino acids have side chains which are polar, but not substantially
acidic. Two of these have alcohol functions and two have amide functions.
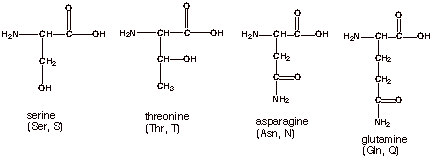
q
4 Naturally occurring
amino acids have acidic side chains, two of which contain an additional
carboxyl group, and so are especially acidic. The other two contain either a
phenolic hydroxyl group or a sulfhydryl group, which are less acidic than the
carboxyl group.
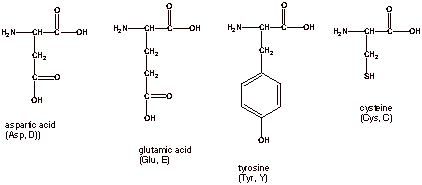
q
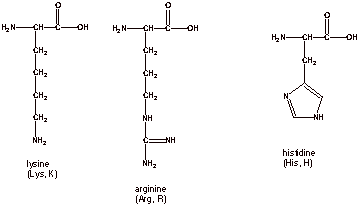
There are 3 naturally
occurring amino acids which have basic side chains, so in an overall sense they are basic amino acids.
The Zwitterionic Structure of Amino Acids
q
In our earlier chapter
(first semester) on acidity and basicity, we learned that carboxylic acids are
strong enough acids to protonate amines very extensively (or to put it
differently, amines are strong enough bases to deprotonate carboxylic acids.
The result is the formation of a salt containing a carboxylate anion and an
ammonium cation.
q
Since both carboxyl and
amino functions are present in amino acids, it should not be surprising that
these two functional groups tend to react with each other in the same way,
giving what can be called an “internal salt”, which contains both
the carboxylate anion and an ammonium cation function.
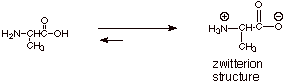
q
The equilibrium lies far
to the right, as in the case of the neutralization of a carboxylic acid by an
amine, so the predominant structure of an amino acid is the zwitterionic
structure. As a “salt-like” substance, it is water soluble and has
a high melting point.
In Acidic
Solution.
In
acidic solution, the carboxylic acid function of the zwitterions is protonated,
thus forming a cationic structure which has an ammonium ion functional group.
This is the conjugate acid of the zwitterions (and also of the neutral,
uncharged, form of the amino acid, by protonating the amino group).
The predominant form of the
amino acid in substantially acidic solution is therefore the cationic form.
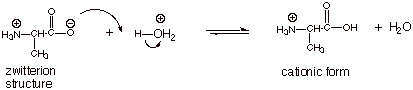
Acidity of the
Cationic Form.
The acidity of the cationic
form of an amino acid is measured by the pKa of the cationic form,
as indicated in the equation below. For all of the amino acids, this value of
pKa is about 2, i.e., this carboxylic acid if fully three powers of
ten more acidic than a typical carboxylic acid (pKa ca. 5).
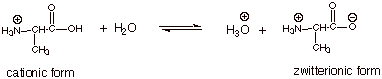
q
This is fundamentally
because the positively charged ammonium function is strongly electron
withdrawing and destabilizes the
carboxylic acid moiety of the reactant (the carboxyl group has a large partial
positive charge on the carbonyl carbon, which is repulsed by the positive
charge on nitrogen). We might also observe, in terms of anion stability, the
the anionic carboxylate moiety in the product zwitterions is stabilized the the
positive charge on the ammonium moiety. Both of these act to enhance the
stability of the cationic form.
In Basic
Solution.
In basic solution, a proton of the ammonium functional
group is removed, leaving an anionic species, which has a carboxylate anion
function. This is the conjugate base of both the zwitterions and the neutral
(uncharged) amino acid.
q
In substantially basic
solution, the predominant form of the amino acid is the anionic form.

Acidity of
the Zwitterion.
q
The zwitterion also has
an acidic function, which is the ammonium substituent. The acidity of the
zwitterion is measured by its pKa, corresponding to the following
equation.

q
The pKa of
the conjugate acid of a typical primary amine is about 10.6, but the pKa
of the zwitterions of a typical amino acid is less than this, about 9-9.5.
q
This means that the
zwitterion is somewhat more acidic than the protonated form of a typical primary amine.
q
That could be somewhat
surprising in view of the presumed stabilization of the reactant side (the
zwitterions) by the attraction between the positive and negative charges. This
would tend to make the zwitterion less
acidic than a typical conjugate acid
of a primary amine.
q
This is undoubtedly a
real effect, but it must be overbalanced by some effect which either produces a
net destabilization the reactant or which stabilizes the product. Most
probably, it is net destabilization of the reactant, i.e., a effect
which counterbalances the plus/minus stabilization effect.
q
We should recall that even in the
carboylate anion, the carbonyl group still has a carbonyl carbon with partial
positive charge (the negative charge of the anion is delocalized over the two
oxygen atoms). So the positive charge on the ammonium substituent destabilizes
the carboxylate anion by repulsing the positive charge on the carboxyl carbonyl
carbon. This positive charge is closer to the ammonium substituent than the
negative charge on the oxygens of the carbonyl group, so the destabilizing
effect is greater than the stabilizing effect.
Isoelectric
Points.
The
isoelectric point of nan amino acid is the pH at which essentially all of the
amino acid exists in the zwitterionic form. At lower pH’s it exists at
least partially in the cationic form and at higher pH’s in the anionic
form.
q
The isoelectric point,
called the pI, is exactly half way between the pKa of the cationic
form and that of the zwitterions.
q
For example, the pKa
of the cationic form of alanine is 2.35, while the pKa of the
zwitterionic form is 9.87. The pI of alanine is therefore 6.11, i.e., one-half
of the sum of 2.35 and 9.87.
q
The significance of the
pI is that at the pI of an amino acid, essentially all of the amino acid exists
in the zwitterionic form, which is overall electrically neutral. The amino acid
therefore does not move toward a positive or negative electrode in
electrophoresis.
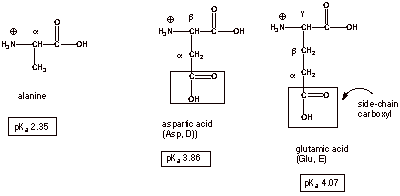
Acidity of
Side-Chain Carboxyl Groups.
q
Aspartic acid and
glutamic acid both have additional, side chain, carboxyl groups, and the acidities of the cationic forms
of these carboxyl groups are also affected by the presence of the ammonium
substituent.
q
In aspartic acid, the
ammonium substituent is rather proximate to the side-chain carboxyl, so the pKa
is substantially lower in comparison to a typical carboxylic acid like acetic
acid. We could say that the ammonium substituent is beta to the carboxyl function. The pKa of this
side-chain carboxyl group is lowered to 3.86.
q
Notice that this is a
much smaller acidifying effect that that the ammonium subsituent exerts on the
cationic form of the main carboxyl (pKa of alanine is 2.35), where
the ammonium substituent is alpha to the the carboxyl group.
q
Finally, the effect is
present, but at a lower level still in glutamic acid (pKa 4.07),
where the ammonium substituent is gamma to the carboxyl group.
Basicity of
the Guanidine Group of Arginine.
The side chain of the amino acid arginine contains a
guanidinyl substituent, which is more strongly basic than any other neutral
functional group, including any amine.
q
The cationic form of the
guanidinyl group of arginine has a pKa of 12.48!
q
This compares to the
conjugate acid of aa typical primary amine which has a pKa of about
10.6.
q
Thus neutral arginine is
about two powers of ten more strongly basic than a typical primary amine.
q
The pKb of
arginine would be 14.00 – 12.48 = 1.52, corresponding to a relatively strong base.
q
The unusual basicity of
arginine is the result of an extraordinarily powerful resonance stabilization,
corresponding to three virtually equivalent resonance structures.
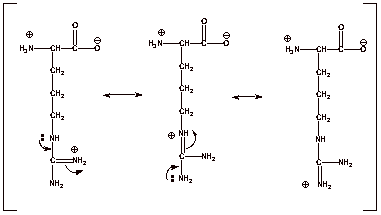
q
Note that the positive
charge is distributed virtually equally over three different nitrogen atoms.
Basicity of
the Imidazole Group of Histidine.
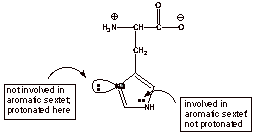
The
imidazole (heteroaromatic) ring of histidine has two different types of
nitrogen atom. Which one is more basic, i.e., which one is more readily
protonated to generate the cationic form?
q
Recall (as in pyrrole)
that a nitrogen atom which contributes two electrons to an aromatic ring having
six electrons in a cyclic system is not readily protonated because that would
destroy the aromaticity and the resonance stabilization would be lost.
q
Therefore the nitrogen
which is formally double bonded to carbon and which has its electron pair in
the trigonal plane of the pi system (not involved in the aromatic system) is
protonated.
Amide/Peptide Functional
Group: Structure and Bonding
The
resonance stabilization of the amide functional group has a major effect on not
only its thermodynamic stability, but upon its structure and its ability to
hydrogen bond.
q
Recall that the amide
function is more resonance stabilized than either the ketone, carboxylic acid,
or ester functions, because the positive charge in the third resonance
structure is more favorably disposed than upon the oxygen atom of a carboxylic
acid or ester function.
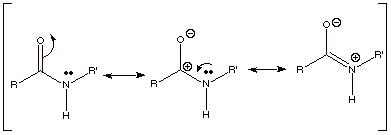
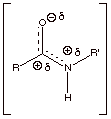
q
We have also previously
noted that owing to the third resonance structure the C-N bond has extensive
double bond character, shortening this bond and making it difficult to rotate
around it (as in the case of a C=C pi bond).
q
For this reason, the
interconversion of trans and
cis conformers exist and their interconversion is now relatively
difficult. They can even be considered as isomers. The cis isomer is
sterically hindered, and so is somewhat less thermodynamically stable than the trans
isomer.
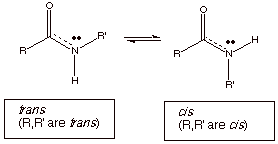
q
We note also that the
oxygen atom of the amide function has even more partial negative charge than in
an ester or carboxylic acid function. It is therefore an exceptionally god
hydrogen bond acceptor.
q
Similarly, the N has a
substantial partial positive charge, making the attached proton relatively
acidic. It is therefore an exceptionally good hydrogen bond donor. We will shortly see how hydrogen bonding between amide
functions determines the secondary structure of the peptide or protein.
Planarity of
the Amide System.
We have previously noted that, unlike the nitrogen
atom of amines (which is sp3 hybridized), the nitrogen atom of
amides is sp2 hybridized.
q
This is in order to
maximize pi bonding (resonance stabilization), which is optimized when the
unshared pair is in a 2p orbital.
q
So strong resonance
stabilization is also the reason for the unusual hybridization state of
nitrogen in amides.
q
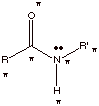
Since the carbon atom of
the carbonyl is planar and sp2 hybridized, and the nitrogen is also
planar and sp2 hybridized, and since the trigonal planes of both the
carbon group and the amine nitrogen must be coplanar (coincident) in order to maximize the pi bonding
implied by resonance stabilization, the amide function has six of its atoms in a common plane. The amide function
adds much planarity to a molecular system.
q
The six atoms of an
amide function which are coplanar are indicated below with asterisks. Note that
in respect to the R and R’ groups, it is only the carbon at of R or
R’ which is directly bonded to the carbonyl carbon or the nitrogen,
respectively, which are constrained to be planar.
The Synthesis of
the Amide Linkage.
We have previously noted that, since the amide linkage
is thermodynamically more stable than a carboxylic acid or ester linkage, the
synthesis of amides from carboxylic acids (or potentially esters, too) is
thermodynamically favorable.
q
However, we have shown
that neither specific acid nor specific base catalysis is available to
accelerate the reaction between a carboxylic acid and an amine.
q
Strong heating (with
help from general acid catalysis) is available to form simple amides, but this
treatment is far too forceful for forming more complex amides, such as
peptides.
q
An appropriate method
for forming amides from amines and carboxylic acids efficiently and under very
mild conditions is, however, available, viz. catalysis by diimides,
specifically (usually) dicyclohexylcarbodiimide (DCC).
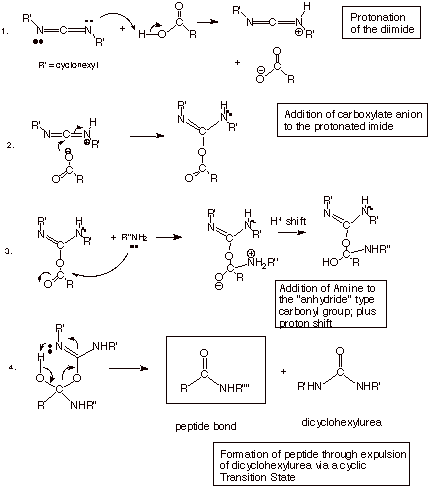
q
The mechanism for the
joining of an amine function to a carboxylic acid function under diimide
catalysis is shown below.
Peptides: Synthesis and
Nomenclature
Dipeptides. The simplest prototype of a peptide would contain two amino acid units bound together through an amide or peptide type bond. The structures and names of two different dipeptides which could be formed from one molecule of glycine and one of alanine, depending upon which of the amino acids supplies the amine function and which supplies the carboxylic acid function, are given below.
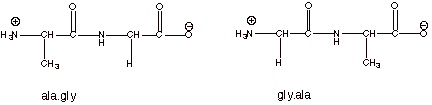
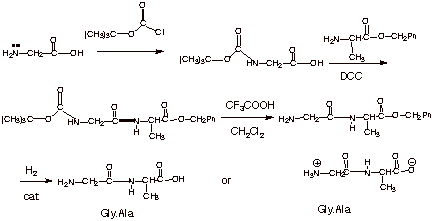
Selective Synthesis. The synthesis of a specific polypeptide or even a dipeptide presents s challenge with regard to selectivity. We can readily see that if we should try to combine one mole of glycine and one of alanine by means of DCC (or any other method of catalysis) can and will obtain not only both of the mixed dipeptides shown above, but also glyclglycine and alanylalanine (four products). If we wish to obtain a single dipeptide, e.g., gly.ala, we will have to arrange it so that the carboxyl group of glycine is used to form the dipeptide bond with the amino group of alanine. Or, to put it another way, we will have to arrange it so that the amino group of glycine is not reacted, and that the carboxyl group of alanine is not reacted. This can readily be done, however by a protecting group strategy. This strategy is illustrated below as a synthetic sketch.
q The amino group of glycine is protected as a special type of amide function which can be readily de-protected. We are taking advantage of the fact that in an amide, the nitrogen unshared pair is principally involved in resonance with the carbonyl group, so that the amide nitrogen is not very nucleophilic. The amide linkage is formed by reaction of the amino group with an acid chloride, tert-butoxycarbonyl chloride.
q The protecting group is called a Boc group.
q The carboxyl group of alanine is then protected as an ester function, by reacting it with an alcohol. The specific alcohol usually chosen is benzyl alcohol, again based upon its ease of subsequent removal.
q The appropriately protected glycine is then combined with the protected alanine in the presence of DCC. The only dipeptide which can result is gly.ala.
q The protecting groups are then removed, the benzyl ester by catalytic hydrogenation and the boc group by treatment with trifluoroacetic acid in dichloromethane solvent.
q Note also that if a tripeptide or higher peptide is desired, either one of the protecting groups could be removed selectively, thus allowing combination with a new protected amino acid at either the amine or carboxyl terminus.
The
Boc Protecting Group.
The tert-butoxycarbonyl
protecting group is an extremely useful and important means of protecting the
amine function of an amino acid.
q
This protecting group is
installed in one of the common ways of generating amide functionality under
mild thermal conditions, viz., reaction of an appropriate acid chloride (highly
reactive and thermodynamically relatively high in energy) with the amine
function.
q
The resulting functionality
is fundamentally an amide type function, albeit of a specific type. As an
amide, the nitrogen atom is far less basic and nucleophilic than an amine
function, so that the amine is protected from reaction.
q
The de-protection of the
Boc protected amine (removal of the Boc function) is equally important. Of
course amides are readily hydrolyzed to carboxylic acids and amines by either
acid or base promotion (not catalysis). However, the aqueous acid or base would
also causes the newly formed peptide link (also an amide) to be hydrolyzed.
q
The Boc protecting group
is used because it can easy be removed under non-aqueous conditions, thus
avoiding the possibility of hydrolysis of the peptides bond or bonds.
q
The mechanism for
removal of the Boc protecting group is given below. Note the importance of the tert-butyl group is that this is an SN1 like
process.
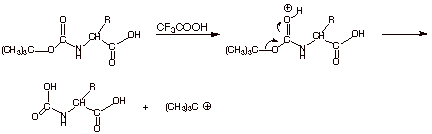
q
Finally, the resulting carbamic
acid derivative is unstable with
respect to decarboxylation, producing carbon dioxide and liberating the free
amine function. This happens spontaneously and quickly.
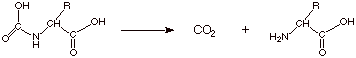
Merrifield Solid Phase Peptide Synthesis
It would be highly inefficient and laborious to try to construct a polypeptide having many amino acids units in the previously described manner in which first the dipeptide is made and isolated and purified, the latter converted to a tripeptide which is isolated and purified and then subjected to conversion to a tetrapeptide, etc. However, the basic concepts of protection and selective incorporation of a desired amino acid can be efficiently employed using what is know as a “solid phase synthesis”, in particular the Merrifield solid phase synthesis.
q The main advantages of the Merrifield approach are that the intermediate oligopeptides need not be isolated and purified and that the method can be automated.
q The basic approach is again illustrated by the synthesis of ala.gly. Whatever the C-terminal amino acid in the target polypeptide is, this amino acid—with the amino group Boc protected—is first attached to a polymer or resin. The polymer is typically polystyrene to which chloromethyl groups have been attached.
q The Boc-protected C-terminal amino acid is bonded to the resin by means of an SN2 displacement of the chloride ion by the carboxylate ion of the cesium salt of the boc-protected C-terminal amino acid.
q The Cs salt is used because it is more reactive than the sodium or potassium salt (see if you can understand why), and because it is more soluble in the non-aqueous solvent (dichloromethane).
q In the laboratory, the Merrifield resin might be placed in a burette or chromatography column or other column and first rinse with pure solvent. Then a dichloromethane solution of the Cs salt allowed to run down the column and react with the resin. The column is then washed with pure solvent as a very simple means of purifying the functionalized resin, i.e., removing the excess Cs salt.
q With the C-terminal amino acid now in place on the resin, the amino group can now be liberated or de-protected by removal of the Boc protecting group (trifluoroacetic acid/dichloromethane). Again, the solution is allowed to run through the column, and after wards the excess of trifluoroacetic acid is removed by washing the column with pure solvent.
q At this point, the next amino acid can be linked to the free amino group of the funcitonalized polymer by running through the column a solution of the Boc-protected amino acid and DCC, followed again by washing the impurities away with pure solvent. In our case the second amino acid added is gly. In the general case, it would be the amino acid next to the C-terminal amino acid. That is, the polypeptide is built up from right to left, as the peptide structure is usually represented (N-terminal on the left; C-terminal on the right).
q If a tripeptide or a higher peptide is desired, the sam procedure can be followed repetitively, i.e., liberating the new amino function with trifluroacetic acid and then adding the new amino acid in the Boc-protected form. Repetition of these two simple states, each followed by washing of the column, can be used to build up higher polypeptides.
q In our case, the dipeptide synthesis is finished. So we simply need to remove the protecting groups. The last Boc group is removed via trifluoroacetic acid treatment, and then the finished product is removed from the resin by cleavage of this benzyl-type ester with HF/dichloromethane.
Basic Elements
of the Primary Structure of Peptides.
We
have previously noted that the strong amide-type resonance stabilization of the
peptide bond causes six atoms to be coplanar. If we start at the alpha carbon
to the carbonyl group, these six atoms include the alpha carbon, the carbonyl
carbon, the carbonyl oxygen, the nitrogen, the hydrogen atom attached to the
nitrogen and the next alpha carbon, which is also bonded to the nitrogen atom.
This does not mean, however, that a polypeptide chain is complete planar,
because the common plane of one set of six atoms does not have to be and is not
coincident with the common plane of the adjoining set of six atoms.
Essentially, this is because the bonds from the alpha carbon, which is sp3
hybridized, to both N and the carbonyl C are free to rotate.
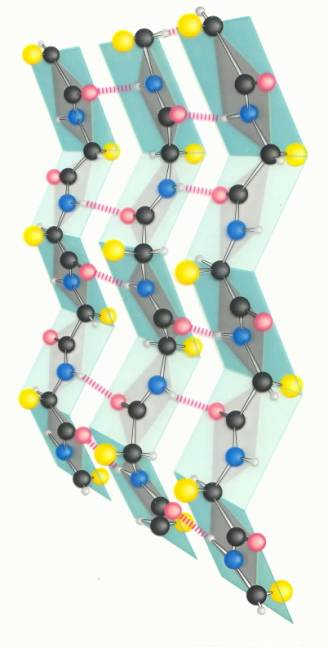
q The illustration above of a part of the beta sheet structure of a polypepide illustrates the pleating which is observed in this type of polypetide structure.
q We not also that the hydrogen-bonding which holds the sheets togethers is of the intermolecular type.
q The R groups of the amino acid residues alternately project upward and downward along the chain, as do the carbonyl functions.
q The other familiar peptide structure is the helical structure, in which the hydrdogen bonding is intramolecular.