SYNTHETIC
POLYMERS AND POLYMERIZATION
A. Polymerization: Cationic Polymerization
We
have seen that the addition of a proton to an alkene linkage gives a
carbocationic species, which is a strong electrophile. Such carbocations are
highly reactive and quickly react with available nucleophiles. In the case of
hydrogen chloride addition, the carbocations react with chloride ion as the
nucleophile, to yield the addition product, an alkyl halide. When there is no
strong nucleophile present and when the solvent is even weakly nucleophilic
(water, alcohols, etc.), the carbocation reacts with the solvent nucleophiles
to give an alcohol or an ether.
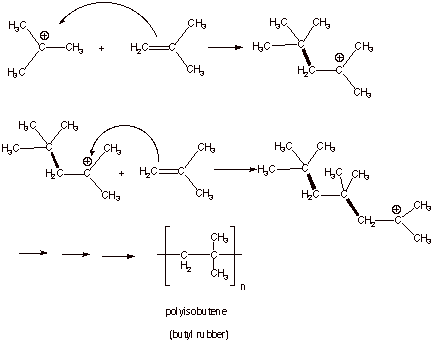
Consider
the case where the solvent is not nucleophilic and there are no other strong
nucleophiles present in abundance. If the alkene is present at high
concentration, the alkene can act as a nucleophile toward the carbocation, forming a dimeric species containing a new
carbon-carbon bond and also a new carbocation center. This process is
illustrated below for the case of isobutene. When this second (dimer)
carbocation reacts with another molecule of isobutene, still another C-C bond
is formed, giving a trimeric species. This can continue until a large number
(n) of isobutene molecules is incorporated into the final product, which is
called a polymer, specifically polyisobutene.
q
This kind of
polymerization is called cationic polymerization, because it involves a chain process which is
propagated by carbocation intermediates.
q
Cationic polymerization
of simple alkenes is especially efficient for alkenes which form relatively
stable carbocations (note the tertiary carbocation intermediate) and which have
one double bond terminus of the alkene unsubstituted, so as to minimize steric
effects in the TS for the addition reaction.
q
Thus, isobutene is
especially amenable to cationic polymerization, but other alkenes with electron
donating groups (oxygen and nitrogen-based functionalities) are also efficiently polymerized.
q
Polymer
Nomenclature: There are several methods
of naming polymers. The simplest method is simply to place the name of the
starting alkene (called the monomer)
in parenthesis and add the prefix poly, as shown above for polyisobutene.
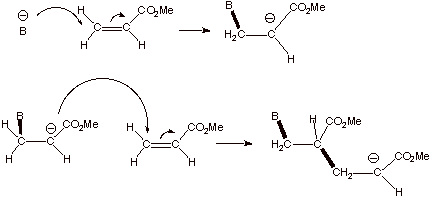
B. Anionic Polymerization
In
contrast to cationic polymerization, anionic polymerization is relatively
efficient when the intermediate anionic species is relatively stabilized. Since
alkyl groups are donor groups, simple alkenes are not polymerized efficiently
by the anionic method. An example of an alkene which is readily polymerized
anionically is methyl acrylate, an alkene which has a strongly electron
withdrawing ester function (carbomethoxy). The intermediate anions will
(hopefully) be recognized as ester enolate anions.
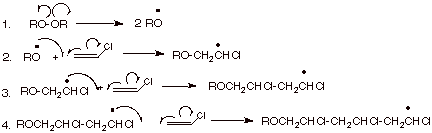
Radical
Polymerization.
You may recall that radical centers are stabilized by either electron donating or withdrawing groups, and especially by conjugating groups. Consequently, radical polymerization is one of the more general methods of polymerization. In fact, even ethylene is readily polymerized to give the familiar polymer poly(ethylene). The mechanism of radical polymerization is illustrated below for the polymerization of chloroethene (vinyl chloride).
q
The repeat structure of
the final polymer is written as:
![]()
q
As is typical for
initiating radical reactions, an initiator, usually a peroxide is employed.
This is because the O-O bonds of peroxides are relatively weak and easily
cleaved homolytically under mild thermal conditions (e.g., 60 – 80 o
C).
c.
[2 pts] Draw the repeat structure of the polymer and provide its formal name.
q
The formal name is:
poly(vinyl chloride) ; note that the parentheses are necessary because the
monomer name is compound.
Polymer
Regiochemistry. Consider the three
possible structures of the polymer of vinyl chloride given below. The
difference between the three structures is essentially a difference in the regiochemistry of the addition reaction. We can
analyze the different regiochemistries illustrated below by considering the two
non-equivalent carbons of the alkene double bond as head and tail respectively.
It doesn’t matter which we consider the head and which the tail, but
arbitrarily let’s consider the carbon bearing the chlorine substituent as
the head and the unsubstituted carbon as the tail. Then we can see that in the
case of A, the tail carbon of on monomer is bonded
to the head carbon of the other and conversely. This is called head-to-tail
regiochemistry. In poly(vinyl chloride) essentially all of the C-C bonds are
formed this way. It is highly regiospecific for the head –to-tail
regiochemistry. Structure C represents a polymer formed by bonding the tail of
one monomer unit to the tail of the other, and also the head of one monomer
unit to the head of another. This is called head-to-head regiochemistry and it
is not observed in this polymerization. Finally, structure B illustrates a
hypothetical polymer which is formed non-regiospecifically, i.e., some of the new C-C bonds are formed in a
head-to-tail manner and some by a head-to-head regiochemistry.
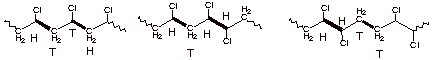
A B C
.
q
The basis for the
preference for head-to-tail regiochemistry is revealed by examining TS models
for the addition step.
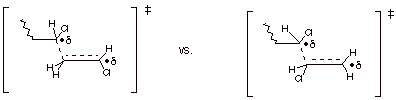
This has radical character This
has primary radical character
stabilized by a chlorine
substituent
It is therefore preferred
over primary
radical character.
q
It should also be
noticed that steric effects play an
important role in determining the preferred regiochemical sense. The second
transition state model has the two more highly substituted alkene carbons
bonding to one another, which represents a higher level of steric repulsion
than in the first TS.
q
The stabilization of the
radical site by the chlorine substituent is based upon three electron bonding.
A molecular orbital diagram is
illustrated below.
![]()
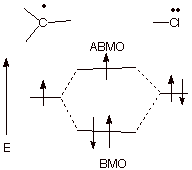
q
As we have noted
previously, when the BMO is double occupied and the ABMO is singly occupied,
there is a net stabilizing effect, termed three electron bonding.
Polymerization
of other Vinyl Monomers. Recall that the vinyl group is CH2=CH-
.
q
Most monomers which are
efficiently polymerized by radical (or other) means are of this type, i.e.,
they have a single substituent replacing one hydrogen of ethene. This minimizes
steric effects in the TS for the addition reaction. Here are a few well known
examples.
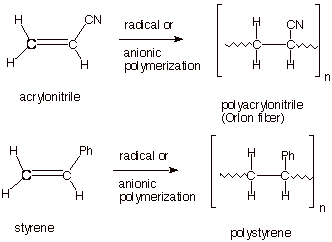
q
A few monomers have two
substituents upon the same carbon (geminal substitution), but it is rare for a
monomer which has substituents on both carbons to be efficiently polymerized by
these means. The basis for this is steric repulsions (see the TS for the
hypothetical head-to-head polymerization of vinyl chloride). Illustrated below
are exceptions to this generality.
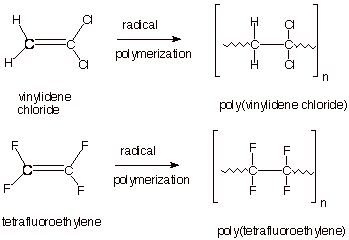
q
The polymerization of a
tetrasubstituted monomer is especially unique. It is possible only because: (1)
the fluorines, though larger than H, are relatively small substituents and (2)
the presence of four very strongly electron withdrawing fluorines on the alkene
double bond strongly destabilizes the pi bond and makes it highly reactive.
q
Although vinylidene
chloride can be polymerized, it is best known for its copolymer with vinyl
chloride (Saran wrap). A copolymer is a polymer formed by incorporating two
different monomers into the polymer chain.
THE POLYMERIZATION OF ETHENE.
As
noted previously, even ethene can be polymerized via the radical method. However, as we will see below,
the polymer formed has a somewhat unexpected structure which contains some
branches at various points along the polymer backbond.
q
We note that the
chain-carrying radicals in the polymerization of ethene are of the primary
type, and therefore are highly reactive.
q
In many cases they add
to another molecule of ethene, as would ordinarily be expected for radical
polymerization.
q
However, some of these
intermediate primary radicals abstract a hydrogen atom from nearby points along
the polymer chain, thereby forming a more stable, secondary radical.
q
We have learned
previously that radicals typically are able to undergo abstraction reactions
and especially hydrogen abstractions. This is particularly the case when the
reaction is intramolecular, as
shown in the mechanism given below, and when the resulting radical is more
stable.
q
One of the favored spots
for hydrogen abstraction is illustrated below, but there are other possible
locations. The particular spot illustrated is favored because the TS for the
abstraction is essentially a six-membered ring.
q
This intramolecular
hydrogen abstraction reaction is termed “backbiting”.
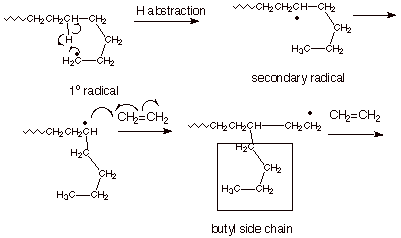
q
The resulting polymer
has butyl (and other) groups as branches along the chain.
q
The presence of branches
on the polymer has a substantial effect upon the physical properties and uses
of the resulting polymer, as we shall see further on.
q
Note that in the
polymerizations of vinyl chloride, styrene, acrylonitrile and virtually all
other monomers, the intermediate radical is much more stable than a primary
radical, so branching does not
occur in these polymers.
Zeigler-Natta
Polymerization.
A fourth and fundamentally different mechanism for polymerization is the so-called Ziegler-Natta polymerization. In this polymerization mechanism, neither carbocations, radicals, nor carbanions are involved. The key intermediates involved here are organometallic compounds. The mechanism for this polymerization type is illustrated below for ethylene.
q
Since no reactive intermediates such as radicals are
involved in Z-N polymerization, no backbiting is observed. The polymer obtained
from ethene is straight chain (i.e., unbranched) polyethylene.
q
Note that the R2Ti groups are end
groups in the polymers, and make little difference ot its properties. They can
be removed and the titanium recovered if desirable.
q
Straight chain (Z-N) polyethylene is a somewhat more
rigid and higher melting polymer
that the branched chain polyethylene generated by radical polymerization.
q
The former is often called high density polyethylene
(HDPE) and the latter low density polyethylene (LDPE). The unbranched chains
fit much more snugly into the solid state lattice, thus requiring a higher
temperature to soften the polymer.
q
The uses of these two types of polymers are therefore
quite different. About 50% of the polyethylene made in the U.S. is of each
type.
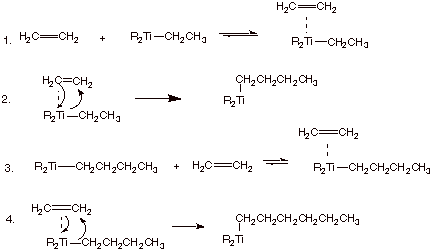
Other Uses of
Ziegler-Natta Polymerization.
Polymer
Stereochemistry. Besides providing the only route to unbranched polyethylene,
the invention of Z-N polymerization has provided solutions to other challenges
in polymer chemistry. One of these lies in the area of polymer stereochemistry.
q
In the polymerization of
vinyl monomers, there exists a stereochemical issue which we have not
previously discussed. The question relates to the relative stereochemistries at
the secondary carbon atoms of the polymer. It is essentially an issue of
diastereoisomerism.
q
The question boils down
to whether the substituents, such as the methyl groups used in the illustration
below of polypropylene, are on the
same side of the polymer chain (shown in the structure on the left) or on
alternating sides of the chain (shown on the right.
q
The left hand structure
is called isotactic and the right
hand one syndiotactic. Both of
these are stereoregular polymers,
in that the relationships between the various methyl groups are
consistent—all on the same side or all alternating.
q
Since stereoregular
polymers have consistent, repeating structures, they fit more snugly in to a
polymer lattice and have higher melting points and greater strength. They are
usually more highly desirable than stereorandom polymers.
q
Radical and other
polymerization methods typically do not furnish stereoregular polymers, i.e.,
some of the substitutents are on the same side and some alternate. These
polymers are stereorandom.
q
Z-N polymerization was
the first polymerization method to provide stereoregular polymers. Both
isotactic and syndiotactic polypropylene have been made using Z-N catalysis.
[Depends upon the specific catalyst]
![]()
isotactic syndiotactic
Conjugate Addition to Dienes to
Give Elastomeric Polymers (Including Rubber).
One
of the challenges of polymer chemistry was to synthesize polymers having
structures identical to (and analogous to) that of natural rubber.
q
Natural rubber is
essentially an addition polymer of isoprene (2-methyl-1,3-butadiene).
[Although, in nature, rubber is not synthesized from isoprene]
q
But there are two major
challenges in polymerizing isoprene to a rubber structure. The rubber structure
has a cis or Z double bond, which
is the less thermodynamically stable isomer.
q
The second challenge is
that the rubber structure corresponds to 100% 1,4 or conjugate addition to the
diene (addition to the diene termini). If you recall, additions to dienes
typically give rise to a mixture of 1,2 and 1,4 additions as a result of
reaction so an intermediate allylic species at either one of the two active
allylic positions.
q
Z-N catalysis elegantly
solves both of these problems and provides a structure identical to that of
naturally occurring rubber.
q
The mechanism for the Z-N polymerization
of the simplest diene, 1,3-butadiene is illustrated below:
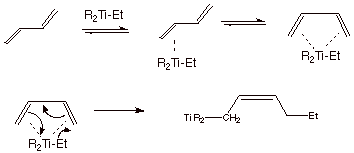
Polymer Repeat Structure:
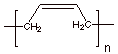
q
The initially formed
coordination complex is formed from the predominant s-trans conformer of the diene. However, rotation around
the central C-C bond of the diene is possible, and this gives an even more
stable coordination complex because both pi bonds of the diene are able to
complex with the titanium atom in the complex of the s-cis-diene.
q
When the addition occurs
in the conjugate manner, the C=C is generated in the cis configuration. The 1,4 addition mode may be favored
because of the six-membered ring TS.
Step Growth vs Chain Growth Polymerization.
All of the polymerizations (except for the Z-N method)
discussed thus far have been chain processes. The growth of the polymer chain
is therefore referred to as chain growth. In many other polymerizations a chain process is not involved, but
many stable intermediates are formed along the way in stepwise fashion. These
are called step growth
polymerizations. A particularly important and familiar example of a step growth
polymer is that of Nylon.
q
Nylon is a copolymer of
a dicarboxylic acid (most commonly adipic acid, i.e., hexanedioic acid) and a
diamine, commonly 1,6-hexanediamine.
q
The first step in Nylon
preparation is simply mixing the reagents, which react (as amins have been
shown to react with carboxylic acids) to form a salt, sometimes called
“Nylon salt”.
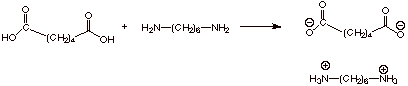
q
This salt is then heated
to form amide bonds between the carboxyl groups and the amine functions (which
are formed by reversal of the nylon salt-forming reaction.
q
An illustration of the
structure of the initial amide formed form one molecule of adipic acid and one
of 1,6-hexanediamine is given below: 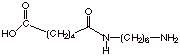
q
Now the importance of
having difunctional comonomers appears. When they have been coupled via the
formation of one amide bond, there still remains a difunctional molecule which
has a carboxylic acid function on one end and an amine function on the other.
This “dimer” can then react at either, and eventually both, ends to
build up the polymer chain.
q
It can do that by
reacting at either end first. Below, we have illustrated the “trimer”
product which would be obtained by reacting the amino end of the
“dimer” (which is the growing polymer chain) with a molecule of
adipic acid.
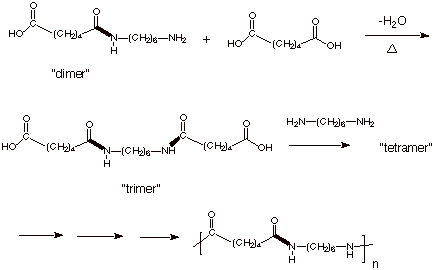
q
We can see from the
nature of the monomers used why this particular Nylon polymer is called Nylon
66. Of course, other dicarboxylic acids and/or other diamines could be used,
but these are two of the most readily available monomers.
q
We can also see from the
mechanism, which proceeds from monomers to dimer to trimer to tetramer, etc,
that this is a step growth polymerization.
Test for Chain Growth vs. Step
Growth Polymerization.
Experimentally,
the nature of a polymerization, whether step or chain growth can be easily
ascertained when we remember the mechanisms of these reactions.
q
In a step growth
polymerization, no polymer of very high molecular weight (high polymer) is
formed until very late in the process, because all of the material has to be
brought up in a stepwise fashion through dimers, trimers, tetramers, etc, etc.
However in a chain process, high polymer is formed very quickly, because the
reactive intermediate proceed all the way to the final polymer product before
stopping.
q
Therefore, if we stop
the polymerization after short reaction times (or better, low monomer
conversions, such as 5-10%; meaning
when only 5-10% of the monomer has been consumed and 90-95% remains), we can
examine whether there is or is not any high polymer formed. It is not so much a
matter of reaction time, but of the fact that the conversion is low.
q
If the monomer consumed
at low conversion is all converted to high polymer, this is a chain growth
process.
q
If only dimers, trimers,
or low oligomers are formed, this is a step growth process.
Polyesters.
Nylon is formed using difunctional comonomers which
form amide linkages. In a similar way, we can combine a dicarboxylic acid and a
diol to give a polyester.
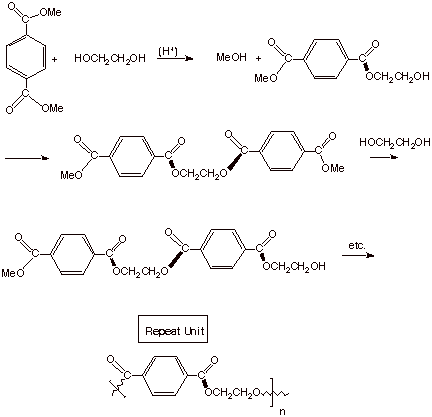
q
Dacron is such a
polyester, formed from terephthalic acid (1,4-benzenedioic acid) and ethylene
glycol (1,2-ethanediol).
q
In practice is is somewhat more
convenient to use a transesterication reaction that an esterification reaction. That is, instead of
terephthalic acid, one uses dimethyl terephthalate and ethylene glycol. In this
case, the condensation product is not water but methanol, which has a lower
boiling point and is more readily removed, allowing the equilibrium to be
pushed toward the product polymer. A transesterification reaction is a reaction
in which one ester function is converted into another ester function.