Chapter 16: Aldehydes and Ketones (Carbonyl Compounds)
The
Carbonyl Double Bond
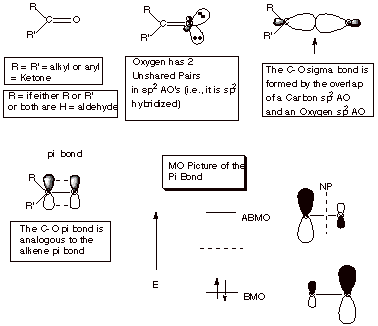
- Both the carbon and oxygen atoms are hybridized sp2, so the system is planar.
- The three oxygen sp2 AO’s are involved as follows: The two unshared electorn pairs of oxygen occupy two of these AO’s, and the third is involved in sigma bond formation to the carbonyl carbon.
- The three sp2 AO’s on the carbonyl carbon are involved as follows: One of them is involved in sigma bonding to one of the oxygen sp2 AO’s, and the other two are involved in bonding to the R substituents.
- The 2pz AO’s on oxygen and the carbonyl carbon are involved in pi overlap, forming a pi bond.
- The pi BMO, formed by positive overlap of the 2p orbitals, has a larger concentration of electron density on oxygen than carbon, because the electrons in this orbital are drawn to the more electronegative atom, where they are more highly stabilized. This result is reversed in the vacant antibonding MO.
- As a consequence of the distribution in the BMO, the pi bond (as is the case also with the sigma bond) is highly polar, with the negative end of the dipole on oxygen and the positive end on carbon. We will see that this polarity, which is absent in a carbon-carbon pi bond, has the effect of strongly stabilizing the C=O moiety.
-
![]()
Resonance
Treatment of the Carbonyl Pi Bond
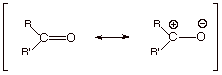
1. Note that the ionic structure (the one on the right
side) has one less covalent bond, but this latter is replaced with an ionic
bond (electrostatic bond).
2. This structure is a relatively “good” one,
therefore, and contributes extensively to the resonance hybrid, making this
bond much more thermodynamically stable than the C=C pi bond, for which the corresponding ionic structure is
much less favorable (negative charge is less stable on carbon than on oxygen).
3. The carbonyl carbon therefore has extensive carbocation
character, while the oxygen has
extensive oxyanion character (or alkoxide character). The corresponding
canonical structure for an alkene pi bond has carbanion character, which is much less energetically favorable than oxyanion
character.
RELATIVE PI BOND STRENGTHS OF THE CARBONYL AND ALKENE PI BONDS
q The bond dissociation energy, D, of an alkene type pi bond
(C=C) is ca. 65 kcal/mol.
q The bond dissociation energy, D, of a carbonyl type pi bond (C=O) is ca. 85 kca/mol.
q The polarity of the carbonyl pi bond is directly responsible for the enhanced bond strength.
q Very Important: The carbonyl pi bond is stronger, it is also more reactive kinetically, because of the carbocation and alkoxide character, the
same feature which makes this bond thermodynamically stronger. The consequence
of this is that reactions at the carbonyl pi bond are often faster than those at the alkene pi bond, but the equilibrium
constant is much less favorable (they do not necessarily proceed to the right).
RELATIVE STABILITIES OF
ALDEHYDES AND KETONES
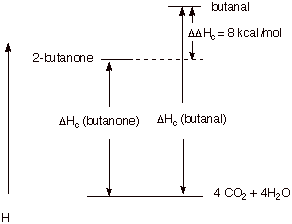
Consider the following isomeric carbonyl compounds:
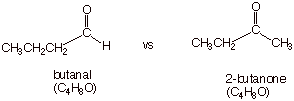
q The equation for the heat of combustion of each is the
same:
C4H8O +
11/2 O2 ® 4 CO2 + 4 H2O
q The heat of combustion of butanal is 8 kcal/mol greater
than that of butanone. Therefore, butanone is 8 kcal/mole more stable,
thermodynamically, than butanal. This is a
reliable generalization for comparisions of aldehydes and ketones.
q The basis for this
stability order is carbocation character at the carbonyl carbon, and the ability of alkyl groups to stabilize carbocation
character. The ketone has two alkyl substituents to stabilize this carbocation character, while the aldehyde
has only one.
q Methanal (formaldehyde), the simplest (one carbon)
aldehyde, has no alkyl groups and is
the least stable of the simple carbonyl
compounds.
q Typically, the least stable carbonyl compounds react most
rapidly, i.e., kinetic rates inversely parallel thermodynamic stabilities. Thus,
the reactivity order is ketones>aldehydes>methanal (formaldehyde).
NOMENCLATURE
ALDEHYDES.
Replace the terminal “-e”
of the corresponding alkane having the same number of carbon atoms with
“-al”.
q The parent (main) chain must contain the aldehyde carbon.
q The aldehyde carbon is always C1, so no locant
number is needed for the aldehyde functionality.
q Examples:
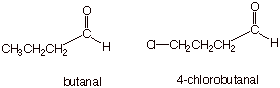
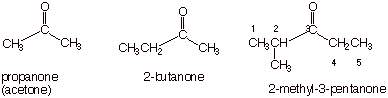
KETONES. Replace
the terminal “-e” of the corresponding alkane with the ketone
suffix “-one”.
q The main chain must contain the ketone carbonyl carbon.
q Numbering of the main chain begins with the end of the main
chain nearest to the carbonyl carbon.
q The position of the ketone carbon must be specified with a
numeric locant (unless there is only one possible ketone having that main
chain).
q In case of ties in regard to the numbering of the main
chain (that is, the carbonyl carbon is equidistant from both chain termini),
numbering proceeds from the end of the main chain closest to the first
substituent.
q Examples:
REACTIONS.
1.
Reactions
with Grignard reagents.
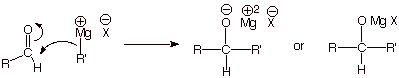
q An analogous reaction, of course,
applies to ketones.
q The reaction is very fast, even at
room temperature, because the carbonyl group has a highly electrophilic
carbonyl carbon,
which combines with the nucleophilic carbon of the Grignard reagent.
q Recall that the C-Mg bond is polar, and the carbon has carbanion character. This is an important
general strategy for forming carbon-carbon bonds: combine a reagent which has
carbocation character with one which has carbanion character.
q Synthetically, the reaction is
used to make alcohols.
The immediate product is the magnesium salt of the conjugate base of the
alcohol, but acidic, aqueous workup provides the corresponding alcohol.
q IMPORTANT: Note that aldehydes give secondary
alcohols, and
ketones yield tertiary alcohols. Addition of Grignard reagents to methanal
uniquely gives primary alcohols.
q Finally, note that Grignard reagents
do not add to alkene pi bonds at all, even though these bonds are much weaker
than the carbonyl pi bonds. This is essentially because the carbons of an
alkene pi bond do not have carbocation character.
q Exercise: Sketch three different
possible syntheses of the tertiary alcohol shown below, using the Grignard
method illustrated above. You may use any Grignard reagent and any carbonyl
compound as starting materials.
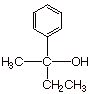
q Exercise: Sketch two different
possible syntheses of the secondary alcohol shown below, using the Grignard
method.
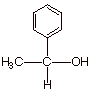
q Sketch a possible synthesis of the
primary alcohol shown below, using the Grignard method of synthesis.
![]()
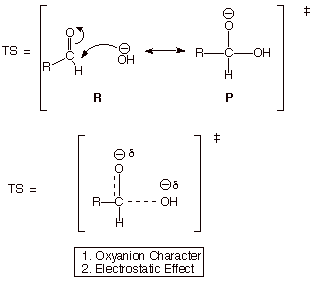
TRANSITION STATE MODEL FOR THE GRIGNARD ADDITION
REACTION.
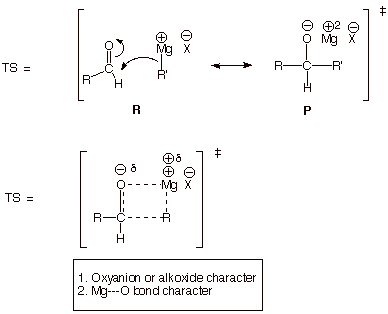
q The alkoxide character derives from the
product (P) character of the TS.
q This alkoxide character is further stabilized by covalent and
ionic bonding to the magnesium ion. The Mg-O bond is a very stable one.
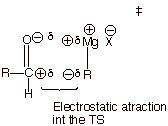
q A final factor which makes this TS an
especially favorable (low energy) one is the electrostatic attraction
between the positively charge carbonyl carbon and the partially negatively
charged carbon of the Grignard reagent. This derives from reactant (R) character in the TS (properly positioned
reactants, of course).
TRANSITION STATE MODEL FOR THE HYPOTHETICAL
ADDITION OF A GRIGNARD REAGENT TO AN ALKENE
q By simply substituting carbon for oxygen
in the transition state model for addition to a carbonyl group (along with
generalized valencies to that carbon), we can obtain a TS model for the
analogous but hypothetical addition of a Grignard reagent to an alkene.
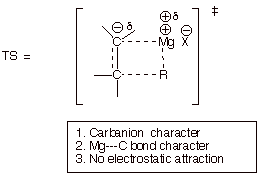
q We use the Method of Competing Transition
States to compare the relative merits of these two reaction (relative rates).
o Carbanion character is much less favorable than oxyanion character.
o Mg-C bond character is less favorable than Mg-O bond
character in the TS.
o Electrostatic attraction is present in the carbonyl addition TS
but is simply missing in the alkene addition TS, because the alkene pi bond is
not polar.
o Consequently, since all of these effects
favor the TS for the addition of Grignard reagents to carbonyl compounds, it is
much more favorable than than the TS for addition to an alkene.
2.
REACTIONS WITH
COMPLEX METAL HYDRIDES HYDRIDE
q The hydrogens of lithium aluminum
hydride and sodium borohydride (see below) are hydridic, since they share a
portion of the negative overall charge of the tetrahydroaluminate or
tetrahydroborate anions. They are therefore nucleophilic.
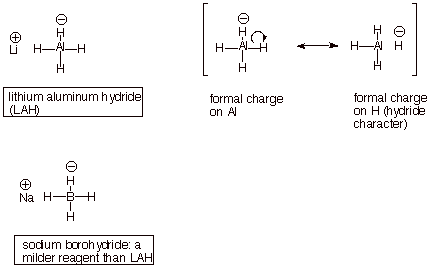
q Just as Grignard reagents provide
nucleophilic carbon, complex metal hydride anions provide nucleophilic
hydrogen. The nucleophilic additions of these hydride reagents to carbonyl
compounds proceed mechanistically very much like the Grignard additions and
also yield alcohols as final products, after aqueous workup.
q The development of a TS model for the
addition of LAH to an aldehyde is shown below:
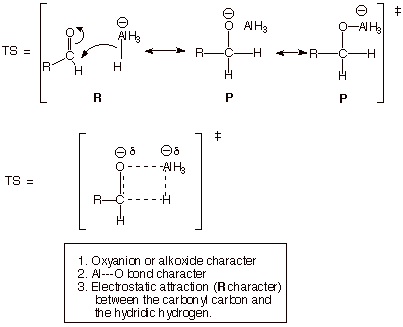
q Since a new carbon-carbon bond is not being added in this case, aldehydes
yield primary alcohols and ketones yield secondary alcohols.
q Exercise: Provide a synthetic sketch,
starting with an appropriate organic halide and using LAH as a reagent, for the
synthesis of the following two alcohols.You may use any appropriate aldehyde or
ketone.
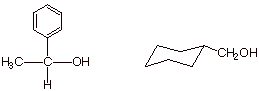
q Explain why a tertiary alcohol cannot be
synthesized using the hydride method.
3.
REACTIONS OF
CARBONYL COMPOUNDS WITH WATER.
THE OVERALL REACTION:

MECHANISM OF
THE BASE CATALYZED REACTION: NUCLEOPHILIC ADDITION (AdN2)
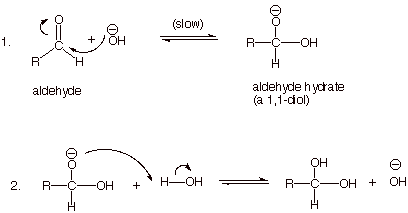
Development
of the Transition State Model for the Slow Step
q The first step is termed
“slow”, but not “rate determining” because it is at
least partially reversible. Recall that a rate-determining step has a rate
which is exactly equal to the rate of formation of the final product. This cannot be the case for a
reversible or partially reversible step, because in this case some of the product of the first step
doesn’t go on to the final product, but reverts to the starting materials
(the reverse of step 1). Therefore the rate of final product formation is less
than the rate of the first (forward) step. Nevertheless, this step is the
slowest step of the reaction and its rate most strongly effects the rate of
product formation. That is why we consider the TS for this step, in loose
analogy to the TS of a rds.
q Note: Hydroxide ion is re-formed in
the second step, so the reaction is truly catalytic.
q Hydroxide anion is the nucleophile,
while the carbonyl carbon is the electrophilic site.
q Both the electrostatic attraction between the carbonyl carbon and the
negatively charged nucleophile in the proprerly oriented TS (reactant
character) and the development of oxyanion character in the TS are favorable factors
which cause this reaction to occur at a rapid rate, even though the C=O bond
is a relatively strong one.
q The first step, though fast, is
reversible as a direct result of
the strength of the C=O bond. The latter bond strength also causes this equilibrium
to lie on the side of the carbonyl compound (to the left in the equilibrium, as
written).
q Analogous nucleophilic additions to
the much weaker
pi bonds of simple alkenes do not occur at all (see analogy to the Grignard and hydride reactions), since carbanion
character is far
less favorable than oxyanion character and there is no assisting electrostatic
attraction in the case of addition to a non-polar alkene.
q The problem with addition of water to
alkenes is not the equilibrium position, but the rate (as reflected in the
discussion of TS characters). The equilibrium for hydration of an alkene (as
you may recall from the first semester) lies well to the right, in favor of the
alcohol. Again, the reason for this is the relative weakness of the C=C pi bond. So in both rate and equilibrium,
hydration of an alkene is in sharp contrast to hydration of a carbonyl
compound.
q The second step of the two-step reaction mechanism is a
proton transfer from oxygen to oxygen; such reactions are extremely fast and
reversible.
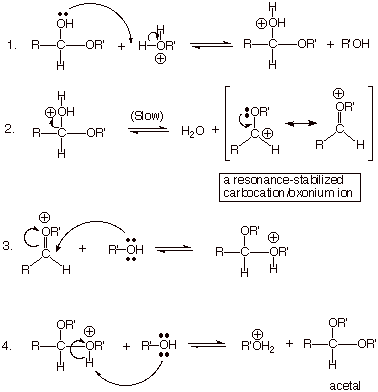
Mechanism of the Acid-Catalyzed
(Electrophilic) Addition of Water to Carbonyl Compounds (AdE2)
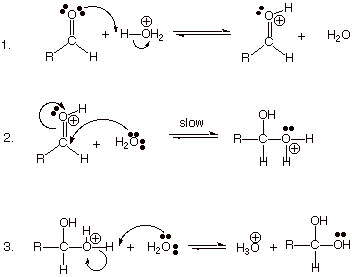
q Note that water is too weak a
nucleophile to add to the neutral carbonyl group at a reasonable rate, but if
the carbonyl group is protonated to provide the conjugate acid shown, the
carbonyl carbon becomes even more electrophilic (it has more partial positive
charge), and even water can add to this.
q The conjugate acid of the carbonyl
compound actually has two resonance structures, one indicating oxonium ion
character and the other carbocation character.
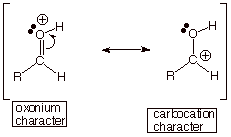
q Although the neutral carbonyl compound
also has carbocation character, the structure which exhibits this character has
charge separation (see below). Therefore this structure is of higher energy and
contributes less to its resonance hybrid than the carbocation structure in the
conjugate acid, which is not charge-separated.
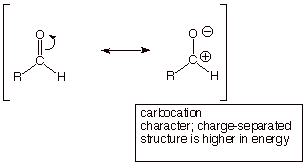
q It is important to again recognize that
the slow step is the
one in which the strong C=O bond is broken (viz., the 2nd step). The
other steps are extremely rapid proton transfers from O to O.
q The original acid, hydronium ion, is
regenerated in the third step, so the reaction is catalytic.
q Interestingly, acid catalyzed hydration
is much faster than the corresponding acid-catalyzed hydration of an alkene
(though the alkene pi bond is weaker). But again, the equilibrium is more
favorable for alkene hydration than for carbonyl hydration.
q In acid catalyzed hydration of an
alkene, the rds is protonation of the pi bond. This requires breaking the pi
bond. In acid catalyzed
hydration of a carbonyl compound, this protonation is quite easy because no
bond has to be broken,
the unshared electron pair on oxygen is a non-bonding pair of electrons. Once
this conjugate acid is formed, it is extremely reactive because it has such
extensive carbocation character. The unshared pair of electrons on oxygen
provides an easy way to facilitate addition to the pi bond.
MECHANISM OF ACID
CATALYZED HYDRATION OF AN ALKENE
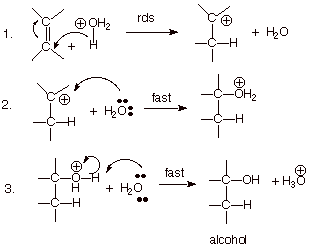
POSITION OF THE
EQUILIBRIUM IN THE HYDRATION OF KETONES AND ALDEHYDES
q The equilibrium between a ketone and
water, on the one hand, and its
hydration product on the other strongly favors the reactant ketone, because of
the strength of the C=O pi bond.
For example, the hydration of acetone, at equilibrium, forms only 0.1%
of the hydrate; 99.9% of the acetone remains unhydrated.
q In the case of an aldehyde, the
equilibrium is somewhat more favorable for hydration than for a ketone, but the
equilibrium still somewhat favors the aldehyde. In neither case does the
reaction go to completion.
q Of the simple aldehydes, only methanal
is more or less completely hydrated in the equilibrium.
q These relative extents of hydration of
carbonyl compounds follow the order of thermodynamic stabilities discussed
previously, viz., ketones are more stabilized than typical aldehydes than
methanal. Thus, alkyl groups stabilize the carbonyl group (via carbocation character) more than they
stabilize the hydrate.
q Again, it is important to note that
hydration of an alkene pi bond goes to completion, whereas the hydration of
carbonyl compounds does not. This accords with the relative thermodynamic
stabilities of the C=C and C=O pi bonds.
HEMIACETAL FORMATION:
ADDITION OF ALCOHOLS TO CARBONYL COMPOUNDS
q The addition of alcohols to carbonyl
compounds follow exactly the same mechanisms as for addition of water.
q The reactions, as in the case of water,
can occur either by an acid-catalyzed or a base-catalyzed mechanism.
q The product hemiacetal, instead of being
a 1,1-diol, is a 1-alkoxy-1-ol, i.e., an ether-alcohol instead of a diol.
q Hemiacetals are so-called because they
are half way toward the formation of an acetal from a carbonyl comound, which
we will consider momentarily. An acetal is a 1,1-diether (a 1,1-dialkoxy
comound).
The Acid-Catalyzed Mechanism for
Hemiacetal Formation.
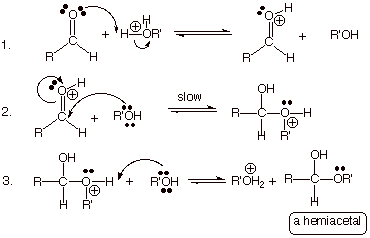
q Note that the only difference from
acid-catalyzed hydration is that R’ replaces one of the hydrogens. The
catalyst, instead of being hydronium ion is the conjugate acid of the alcohol,
since the reaction would be carried out in the alcohol as the solvent.
q Also, the alcohol replaces water is the
nucleophile in step 2 and as the base in step three.
q The hemiacetal product also has one
OR’ group replace one of the alcohol functions.
The Base-Catalyzed Mechanism of Hemiacetal
Formation
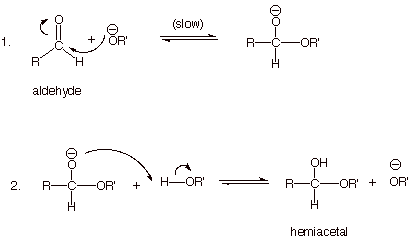
q Note that the nucleophile in step 1
and the base in step 2 is not hydroxide ion, but an alkoxide anion.
ACETAL
FORMATION
q Acetals are formed from hemiacetals
in a strictly acid-catalyzed process. Base catalysis is ineffective in
converting a hemiacetal to an acetal.
q Although hemiacetal formation is an
addition reaction (AdE or and, depending upon whether the mechanism
is acid- or base-catalyzed, the conversion of a hemiacetal to an acetal is a substitution
reaction.
q The overall reaction is replacing an
–OH group with an OR’ group. The mechanism is most similar to an SN1
substitution mechanism.
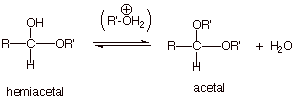
REACTION MECHANISM:
FORMATION OF CYCLIC
ACETALS FROM DIOLS
q The formation of acetals usually
starts with the carbonyl compound and proceeds via a two stage process, the
first (addition)stage yielding the hemiacetal and the second (substitution)
stage converting the hemiacetal to an acetal. Recall that, whereas base
catalysis can accomplish the first stage, it cannot convert a hemiacetal to an
acetal (we will return to discuss the reasons for this momentarily).
q The overall reaction for the
conversion of a carbonyl compound to an acetal (see below) requires two moles
of alcohol.
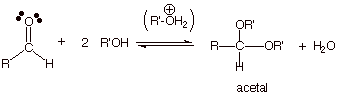
q If, instead, a diol is used to
provide both R’O groups, a cyclic acetal is formed. The most commonly
used diol is the very readily available ethylene glycol (1,2-ethanediol).
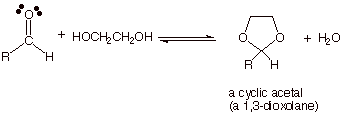
q In comparing these two equilibria
(one using two molecules of a monofunctional alcohol and one using one molecule
of a difunctional alcohol), we can see that in the first case three molecules of reactant are converted
to two molecules
of product, but in the second case two molecules of reactant are converted to two molecules of product.
q This is important because molecules
are free to move about (translate) and this freedom contributes to their
thermodynamic stability through the entropy factor. This freedom is referred to
as translational entropy.
When a reaction takes place which requires the loss of translational entropy,
this is an unfavorable factor with respect to the free energy change and to the
completion of the equilibrium. When 2 molecules of reactant are converted to 2
molecules of product, little or no loss of translational entropy occurs. But
when 3 molecules of reactant are converted to 2 molecules of product, the
translational entropy of one molecule is lost. This is a very significant
factor with respect to the position of the equilibrium.
q As a result , the equilibrium for the
formation of a cyclic acetal is much more favorable than for an acylic acetal.
In particular, the equilibrium for the formation of an aldehyde cyclic acetal
is now favorable to the acetal, whereas it was not in the case of an acylic
acetal. Consequently, the formation of a cyclic acetal in organic synthesis is
much more convenient than forming an acylic acetal.
q In the case of cyclic acetal
formation, the step which is really making the difference is not the addition
step, but the second, substitution step, because this step is essentially
intramolecular. Intramolecular reactions are typically strongly favored by entropy (note
that one molecule is converted to two molecules, thereby increasing
translational freedom).
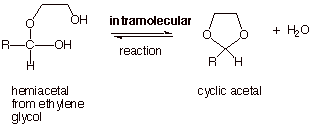
WHY IS BASE-CATALYSIS
INEFFECTIVE IN CONVERTING HEMIACETALS TO ACETALS??
q Recall that hydroxide ion is a
rather poor leaving group,
whereas water is a good leaving group.
q Refer to step 2 of the mechanism for
the conversion of a hemiacetal to an acetal, noting the analogy to an SN1
reaction, with water as the leaving group. In the absence of acid, the leaving
group would have to be a hydroxide anion, which is a poor leaving group.
q It is also important to note that the
converse is alsotrue, that is, that an acetal cannot be converted to a hemiacetal
in base.
q Thus, the acetal function is
especially synthetically useful because it is stable in basic solutions and contains only
two relatively unreactive ether functionalities. On the other hand, it is
readily converted back to the precursor carbonyl compound by acid-catalyzed
hydrolysis. The
mechanism is the reverse of acid-catalyzed acetal formation (start with the
product and read backward in the mechanism).
SYNTHETIC
APPLICATIONS OF ACETALS: THE PROTECTING GROUP STRATEGY
q The ability to smoothly convert
reactive aldehyde or ketone functionality to a relatively inert acetal
functionality is highly useful in organic synthesis. The acetal functionality
is, in effect, a protected carbonyl group.
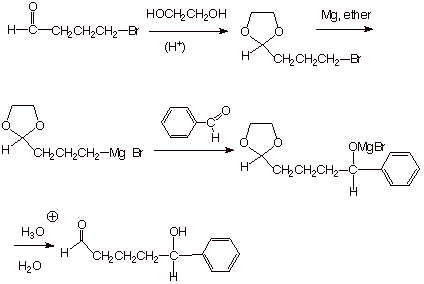
q An excellent example of the use of
the acetal protecting group strategy is the formation of a Grignard reagent
from a bromoaldehyde, as shown below. If the aldehyde functionality were not
protected, this Grignard reagent could not be formed in a synthetically useful
manner, because it would quickly react with a molecule of the aldehyde.
However, if the aldehyde functionality is first protected as an acetal
function, the Grignard can be prepared quite nicely, and the protected Grignard
reagent can then be used for whatever synthetic purpose may be desired. In the
example shown, the Grignard is used to react with benzaldehyde to give, after
hydrolysis, a hydroxyaldehyde.
q Note that there are essentially three
requirements for an
effective protecting group.
o It must be formed in high yield
o In the protected form it must be
unreactive
o The protected functionality must be
efficiently re-converted to the original functionality. This step is called
“de-protection”.
q The de-protection step is very facile
in the case of an acetal, because in the course of acidic, aqueous workup it is
quickly hydrolyzed to the carbonyl functionality.
CONSIDER ANOTHER
EXAMPLE OF THE PROTECTING GROUP STRATEGY:
q In the case of the synthetic
transformation below, the preferred starting material is a keto ester, and it is desired to selectively
reduce the ester functionality to
an alcohol functionality, without affecting the ketone function (the
target molecule retains the ketone function).
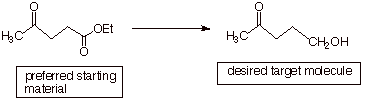
q It is known that lithium aluminum
hydride will efficiently reduce an ester function in the manner desired (see
later), but we also know that it will reduce the ketone function, in this case
to a secondary alcohol. This would not lead to the desired product, if both
functionalities were reduced.
q There is an even more serious
problem. If we were to attempt to use only the stoichiometric amount of the
hydride reagent necessary to selectively reduce one of the functionalities, the ketone would
be the one preferentially reduced, because the ester function is resonance
stabilized (see later), and therefore less reactive than the ketone. (By the
way, LAH is highly reactive, and not very selective, anyway, but any
selectivity would favor reaction with the ketone function.
q We must therefore adopt a protecting
group strategy, as shown below. Again, note that the acidic, aqueous workup
required for LAH reductions is also that required to de-protect the acetal
functionality.
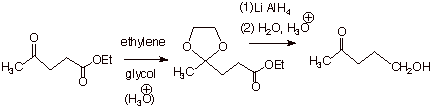
ADDITION OF OTHER CARBON
NUCLEOPHILES TO CARBONYL COMPOUNDS
I.
Additions
of Conjugate Bases of Terminal Alkynes.
q Since terminal alkynes are unusually
acidic, their conjugate bases may be prepared quantitatively by using a
sufficiently strong base. In this connection, sodium amide is an appropriately
strong base, as we learned in the first semester (the pKa’s of
terminal alkynes are ca. 25, whereas the pKa of ammonia is ca. 38).
q These anions are carbon centered
anions (carbanions), and, like Grignard reagents, are highly nucleophilic. They
therefore add in the same manner as do Grignard reagents to carbonyl compounds.
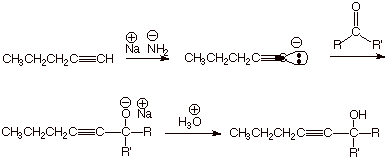
II.
Addition of
Cyanide Ion.
q The cyanide anion is another carbon
centered nucleophile which can add readily to carbonyl functionalities. As in
the case of Grignard reagents and alkynide anions, the special importance of
cyanide anion as a nucleophile is that it forms a new carbon-carbon bond.
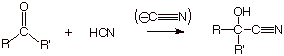
q The reaction only requires a catalytic
amount of the cyanide
anion, since the intermediate alkoxide anion (the conjugate base of the
product) can abstract a proton from HCN, generating more cynanide anion.
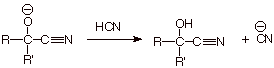
q The CN functionality is called a nitrile group. Nitrile groups are at the same
oxidation state as a carboxylic acid group or an ester group, and can be
readily converted to either one of these
q You may recall from the first
semester’s studies of SN reactions, that the cyanide anion is
a very strong nucleophile.
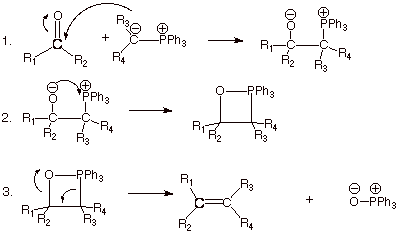
THE WITTIG
REAGENT
q .The so-called “Wittig
reagent” is another reagent containing nucleophilic carbon, and which can
add readily to a carbonyl group. The reagent, as illustrated below, has
extensive carbanion character, and thus is quite nucleophilic.
q This reagent is rather unique in that it
allows the formation of a new doubly bonded carbon. The net result is the conversion of an
aldehyde on ketone to an alkene of choice. The overall reaction is illustrated
below. It can be used to form mono-, di-, tri-, or tetrasubstituted alkenes. A
mixture of cis and trans
alkene isomers are
usually formed.
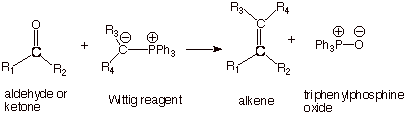
q It is very important to note that the
conversion of a carbonyl function to an alkene double bond is thermodynamically
unfavorable by at least 20 kcal/mol, as we have seen in this chapter. What is thermodynamic “driving
force” which makes this reaction possible? (That is, which provides more
than 20 kcal/mol of negative free energy change?)
q It is, of course, the conversion of the
Wittig reagent to triphenylphosphine. In particular, the Wittig reagent has carbanion
character, but the
phosphine oxide has oxyanion character, the latter being very much more favorable for
stabilization of the negative charge.
Resonance Treatment of the Wittig Reagent.
q The canonical structure written above for
the Wittig reagent places negative charge on carbon and positive charge on the
tetravalent phosphorous atom. Why not move the electron pair of the carbanion
center in between the carbon and phosphorous atom to form a second bond, i.e.,
a pi bond?
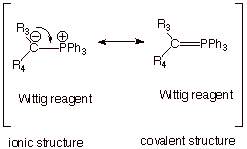
q Actually, for phosphorous this covalent
structure is a valid (i.e., legal) canonical structure, because phosphorous is
a third row element which has vacant 3d orbitals. Although it has already used
all of its valence shell 3s and 3p AO’s to form four sp3
covalent bonds (three to phenyl groups and one to the carbanion carbon), it
still has 3d AO’s, which are vacant and can overlap with the filled
carbon 2p AO which contains the
electron pair. We thus can have a two electron pi bond structure.
q Normally, since the covalent structure has
one additional covalent bond, one would think that this structure would be the
lower energy structure, but in this case, interestingly, the ionic structure
is the one of lower energy.
There are two reasons for this:
q First, the pi bond which is formed is very
weak indeed, because the 3d AO is quite high in energy, and overlap with that
orbital yields relatively little stabilization.
q Secondly, the first structure, although
lacking a pi covalent bond, does have a relatively strong ionic (i.e.,
electrostatic) pi bond.
Of course, ionic bonding can be quite strong.
q Consequently, we usually draw the ionic
structure to represent the Wittig reagent, but you should know that a covalent
structure also contributes to the resonance hybrid, so that carbon
doesn’t have a full unit of negative charge.
q Suppose we had an analogous reagent in
which the phosphorous was replaced by nitrogen? Would the covalent structure be
valid? The answer is NO! Nitrogen is in the second row of the Periodic Table
and the valence shell is the second main shell, which has no d type orbitals.
Preparation of the Wittig Reagent.
q Some Wittig reagents are commercially
available, but most are preferably synthesized prior to use in the laboratory.
There are two steps in the generation of an appropriate Wittig reagent:
q Step 1: Reaction of triphenylphosphine with an
alkyl halide, in an SN2 reaction. Recall that P, as a heavier atom,
is especially nucleophilic, although not very basic (the
“polarization” effect), so these displacement reactions work very
efficiently. This reaction results in the formation of a stable phosphonium
salt (tetravalent
phosphorous is analogous to tetravalent nitrogen of ammonium salts). The simplest case, reaction with methyl
iodide, is illustrated below, but analogous reactions with primary or secondary
halides are feasible.
![]()
q Step 2: Removal of a modestly acidic proton from
the carbon atom alpha to the positively charged phosphorous atom. This proton
is removable because the resulting “carbanion” will be
substantially stabilized by the resulting electrostatic bond present in the
Wittig reagent and to a modest extent by the weak pi bonding present in the
Wittig reagent. Any of a variety of bases can be used, but relatively strong
ones like butyl lithium
are more commonly used.
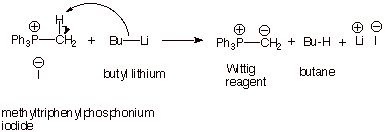
Mechanism of the Wittig Reaction.
AN EXAMPLE:
q Sketch an efficient synthesis of the
following alkene:
![]()
q Answer:
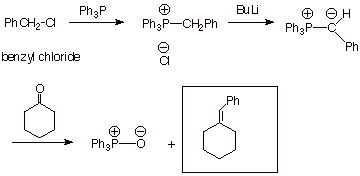
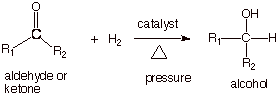
CATALYTIC HYDROGENATION.
q You already know that alkene pi bonds can
be readily hydrogenated to alkanes by reaction with dihydrogen in the presence
of platinum, palladium, or nickel catalysts. These reactions occur under very
mild conditions: room temperature with just 1 atmosphere of hydrogen.
q You also may know that benzene and
aromatic hydrocarbons require more forcing conditions for the hydrogenation of
the highly resonance stabilized, aromatic ring. Forcing conditions refers to
the use of much higher temperatures and pressures.
q In much the same way as alkene pi bonds,
carbonyl pi bonds can be hydrogenated to alcohols. The conditions required,
however are quite different, since the carbonyl pi bond is much more stable
thermodynamically than the alkene pi bond. For the hydrogenation of a
carbonyl group, forcing conditions are required.
q Consequently, a molecule which contains an
alkene pi bond and a carbonyl pi bond can, if desired, be selectively
hydrogenated at the alkene pi bond. In the example shown, the starting material
has an aromatic ring, a ketone function, and an alkene function. Under mild
conditions only the alkene function is reduced.
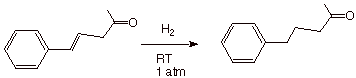
EXAMPLES OF
SYNTHESES USING GRIGNARD REAGENTS
Example 1: Sketch an efficient synthesis of the
target compound shown below using any organic compound having four carbon atoms
or less, and any inorganic reagents, solvents and reaction conditions
necessary.
![]()
Answer:
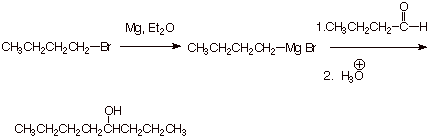
q Note: The use of the three carbon Grignard reagent would require a
five carbon aldehyde, which is not permitted under these ground rules.
Example 2.
Sketch an efficient synthesis of the following target alcohol using any
organic halide and any carbonyl compound.
![]()
Answer:
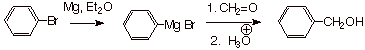
Example 3. Sketch an efficient synthesis of the
following carboxylic acid ( Hint: Use carbon dioxide as a carbonyl component).
![]()
Answer:
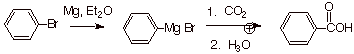
NOTES:
q THE MATERIAL ON ENOLS AND ENOLATES WILL BE
COVERED IN A LATER CHAPTER.
q THE ADDITION OF NITROGEN-CENTERED
NUCLEOPHILES TO CARBONYL COMPOUNDS WILL NOT BE COVERED IN THIS UNIT.