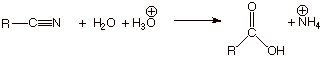CHAPTER 18: FUNCTIONAL DERIVATIVES OF CARBOXYLIC ACIDS
Esters
are considered to be functional derivatives of carboxylic acids in which the
acidic proton has been replaced by an organic group. The synthesis of esters by
the acid catalyzed condensation of a carboxylic acid and an alcohol was
considered at the end of the previous chapter. We can also consider the
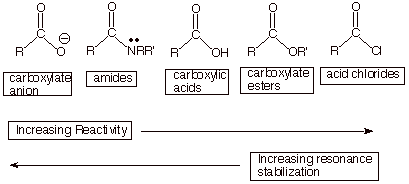
conjugate base of a carboxylic acid, a carboxylate anion, as a functional derivative of the
carboxylic acid. Two additional functional group derivatives will be considered
in this chapter, viz.,
amides and acid
chlorides. Together,
these five classes of organic compounds can also be considered as acyl
compounds, since they all
contain the acyl group (RC=O). Aldehydes and ketones, referred to jointly as
carbonyl compounds, are not considered to be acyl derivatives. The acyl
compounds as a group all contain a carbon atom (the acyl carbon) in the +3
oxidation state, so that their interconversions, which are the special emphasis
of this chapter, are not redox reactions. Carbonyl compounds have the +2
oxidation state of the carbonyl carbon atom.

q
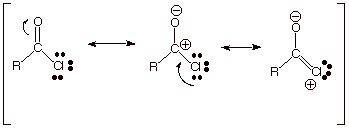
As noted
previously, the carboxylate anion has two equivalent resonance structures and
has the largest resonance stabilization of these acyl compounds.
q
We have also
noted extensively that the resonance stabilizations of the carboxylic acid and
ester functionalities are virtually identical and both very considerable, but
less than for the carboxylate anion.
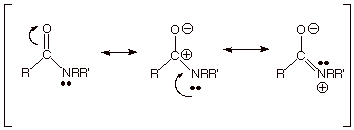
We now want to consider the amide and acid chloride functionalities. First, the amide function.
q
We note that
the nitrogen atom of an amide has an unshared electron pair which can be shared
with the electron deficient carbonyl group (see the third structure,
below). The unshared pair on
nitrogen is less tightly held than that on the corresponding oxygen atom of a
carboxylic acid or ester, because of electronegativity considerations, so that
placing a positive charge on nitrogen is less unfavorable than on oxygen.
Consequently, the third resonance structure for an amide function is a more
major contributor than the third canonical structure for an ester or carboxylic
acid. But of course the
amide does not have two equivalent resonance structures, so it is less highly
resonance stabilized than the carboxylate anion. We can therefore place the
amide function in between the carboxylate anion and the carboxylic acid/ester
groups.
q
We should
also note that the nitrogen atom of an amide is sp2 hybridized, unlike the nitrogen atom of ammonia or
organic derivatives of ammonia (called amines), where the nitrogen prefers the
sp3 hybridization state. The reason for the sudden change in
hybridization is that the sp2 hybridization state places the
unshared electron pair in a 2p AO, so that the pi bonding indicated in
structure three above is optimum. Otherwise the electron pair would be in an sp3
AO on nitrogen. Overlap of this type of orbital with the 2p AO on the carbonyl
carbon would be much less efficient (recall that pi bonding is most efficient
between p type orbitals).
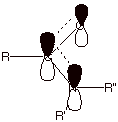
q
As a
consequence, the nitrogen atom of amides is planar, not pyramidal as in amines or ammonia.
Because of the very strong pi bonding between the nitrogen atom and the
carbonyl carbon in amides, the trigonal plane of nitrogen atom is required to
be coincident with the
trigonal plane of the carbonyl carbon atom (also, of course, sp2
hybridized). As a result no less than six atoms are required to be coplanar,
i.e., all of the atoms directly connected to the carbonyl carbon and also to
the amide nitrogen atom.
Specifically, these are the carbonyl carbon, its attached oxygen, the atom (R)
attached to the carbonyl carbon, the nitrogen and the two atoms
(R’,R’’) attached to nitrogen.
Acid Chlorides.
q
In the case
of acid chlorides, the third resonance structure is the least favorable of all
of the acyl compounds, placing a positive charge on the highly electronegative
chlorine atom. Also, the 3p orbital of chlorine is too large to overlap very
efficiently with the much smaller 2p AO on the carbonyl carbon. Consequently,
acid chlorides have the least resonance stabilization and the highest
reactivity of all of the acyl derivatives.
HYDROLYSIS OF ESTERS
q
Recall that
the esterification of a carboxylic acid is completely reversible, so that when
the starting material is an ester, it can be converted to the carboxylic acid
by hydrolysis in aqueous, acidic media. The overall reaction is shown below:
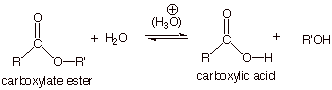
q
Also, recall
that the equilibrium constant is approximately K = 1, so that the reaction only
goes to completion because a large excess of water, used as the solvent, drives
it essentially to completion.
q
The reaction
mechanism is essentially the reverse of that written for esterification, except
that the catalyst is now hydronium ion and the solvent is water (in
esterification, the catalyst is the conjugate acid of the alcohol, which is
often used as the solvent). The mechanism of acid catalyzed hydrolysis of an
ester is written below:
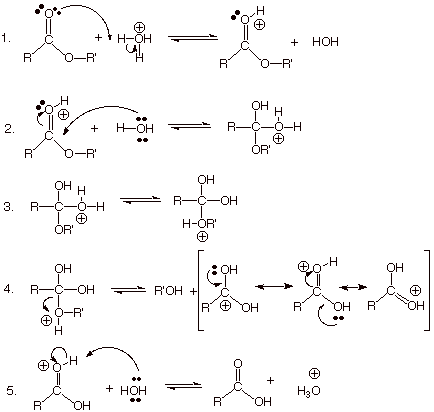
BASE-PROMOTED ESTER HYDROLYSIS (SAPONIFICATION)
q
If you
recall, esterification of a carboxylic acid with an alcohol was not possible,
because the base (presumably one would use the conjugate base of the alcohol,
i.e., an alkoxide ion) reacts with the acidic proton of the carboxylic acid to
yield the highly resonance stabilized, negatively charged carboxylate anion,
while also neutralizing the reactive base. Even if one were to use a large
exces of the alkoxide anion, it would be unable to add to this functionality at
an appreciable rate.
q
In contrast,
an ester does not have the relatively acidic proton, so that the base
(hydroxide ion) adds to the carbonyl group, leading to efficient hydrolysis of
the ester.
q
It should be
noted that the hydroxide ion is consumed in the final step of the reaction, so
that the reaction is not
base-catalyzed, but rather requires a stoichiometric amount of the base. The
reaction is described as base-promoted.
q
Although the
unavoidable consumption of the base requires that we use a mole for mole amount
of sodium or potassium hydroxide, there is also a favorable aspect of this
final, neturalization, step. As noted previously, the equilibrium between a
carboxylic acid and an ester is not highly favorable to either ( K = ca. 1).
However, the final step results in the formation of the highly resonance stabilized
carboxylate anion, providing the thermodynamic driving force for driving the
reaction to completion.
q
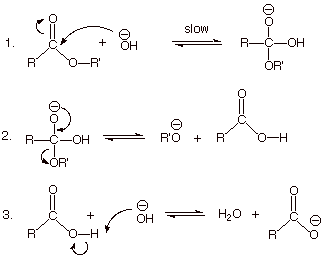
The
mechanism of the base-promoted ester hydrolysis is illustrated below. Note in
particular that the step in which the carbonyl pi bond is broken is the
“slow” step. Also that the reaction is not a direct displacement as
in the case of an SN1 or SN2 reaction, but a stepwise addition/elimination
process.
HYDROLYSIS OF AMIDES TO CARBOXYLIC ACIDS
Amides
can be hydrolyzed to carboxylic acids by either acid-promoted or base-promoted processes. We will consider in detail
only the more common acid-catalyzed mechanism. As background for the acid
catalyzed hydrolysis of amides, let’s consider the basicity of amides.
q
As in the
case of esters or carboxylic acids, there are two basic sites in an amide
function which could potentially be protonated, namely the carbonyl oxygen and
the amide nitrogen. Interestingly, and somewhat surprisingly, protonation of amides occurs
preferentially on the carbonyl oxygen. Surprisingly, because the electron pair
on trivalent nitrogen is typically much less strongly held to the nucleus than
are electron pairs on neutral, divalent oxygen. An example would be the
relatively high basicity of amines such as RNH2,
which are very much more basic than alcohols, ketones, esters, etc.
q
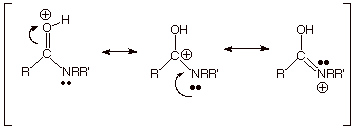
What is
responsible for this dramatic inversion of the relative ease of protonation of
oxygen and nitrogen? The same factor which caused esters and carboxylic acids
to be preferentially protonated on the carbonyl oxygen, rather than the
R’O type oxygen, viz., the carbonyl oxygen-protonated conjugate acid is
highly resonance stabilized (see three resonance structures, including one in
which the unshared pair on nitrogen is used to stabilize the cation).
q
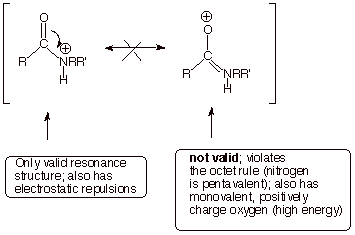
In contrast,
the nitrogen-protonated conjugate acid has no resonance stabilization at all
(there is only one valid resonance structure). The positive charge is also
localized on an atom (N) which is directly attached to a highly electron deficient
carbonyl carbon (electrostatic repulsion).
q
Finally,
note that because of the third resonance structure of the oxygen-protonated
conjugate acid of the amide, amides are substantially more basic than
carboxylate esters or carboxylic acids. This third structure has positive
charge on nitrogen (ammonium character), which is much more favorable than
positive charge on oxygen (oxonium character)
MECHANISM OF ACID-CATALYZED HYDROLYSIS OF AMIDES.
. 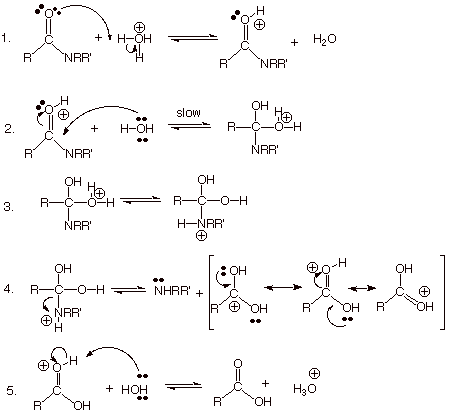
q
NOTE: To
this point, the conversion is from an amide to a carboxylic acid. Since the
amide is far more resonance-stabilized than the carboxylic acid, by the end of
step 5 the equilibrium lies well to the left, i.e., on the side of the starting
amide.
q
However,
there is a sixth step which is highly exergonic and drives the equilibrium to completion.
That step (see below) involves the neutralization of the hydronium ion
regenerated in step 5 by the basic amine formed in step 4. Step 6 is thus an
essential part of the mechanism.
![]()
q
This also
reveals why the reaction is not catalytic, but acid-promoted, i.e., the
hydronium ion formed in step 5 is neutralized by the amine, in step 6.
BASE-PROMOTED
AMIDE HYDROLYSIS.
q
We will not consider the specifics of this mechanism,
but we should understand that the reaction is base-promoted (not catalyzed), and that the reaction
only goes to completion because of a final, neutralization step which is highly
exergonic.
q
This is the
neutralization of the carboxylic acid by the hydroxide anion (just as in the
case of base-promoted ester hydrolysis).
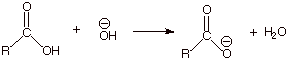
q
The overall
reaction, starting from the amide, goes to completion because the
carboxylate anion has greater resonance stabilization than the amide.
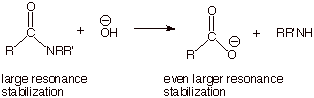
q
Note that
the hydroxide anion is consumed quantitatively in the reaction. It must be used in an
equimolar amount.
q
Another
practical consequence of this neutralization is that in order to obtain the
carboxylic acid, one must do an acidic, aqueous workup in order to protonate the carboxylate
anion, which is the primary product of the reaction.
FORMATION OF AMIDES
Interestingly,
although the formation of an amide functionality from a carboxylic acid is
strongly exergonic and therefore thermodynamically favorable, there is no
simple and effective Bronsted acid or base catalyzed or promoted mechanism for
this reaction. Why not?
q
Bronsted
acid catalysis/promotion is frustrated by a rapid neutralization reaction
between the strong acid and the basic amine required for amide formation. The
use of excess acid is to no avail, because the amine has been fully converted
to the conjugate acid, which is not nucleophilic at all.
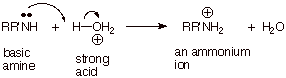
q
Base
catalysis/promotion is also precluded by the neutralization of the carboxylic
acid by the base, giving the unreactive carboxylate anion. This same result
would obtain whether hydroxide ion or any other strong base (such as the
conjugate base of the amine) were used.
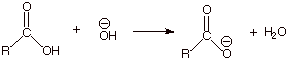
THREE METHODS FOR AMIDE FORMATION
q
Thermal
condensation of a carboxylic acid with an amine.
q
The
carboxylic acid is a weak acid, but at higher temperatures (150 – 200 oC),
it can act as an acid catalyst.
q
In order to
use this method, the compounds (amine and carboxylic acid, as well as the
product amide) must be thermally stable.
q
The reaction
can be accelerated toward completion by removal of water.

q
Reaction of
an Acid Chloride With An Amine.
o
Since acid
chlorides are much less thermodynamically stable than carboxylic acids, or any
of the other acyl derivatives, their reactions with nucleophiles can proceed
rapidly, at much lower temperatures (room temperature) and without the need for
catalysis.
o
This is a
convenient laboratory means for preparing either amides or esters.
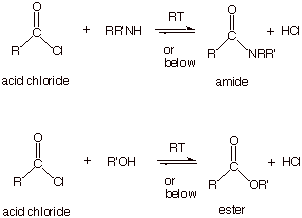
q
Condensation
of a Carboxylic Acid with An Amine via Carbodiimide Catalysis
o
Many
compounds (carboxylic acids or amines) are not thermally stable enough to
survive the elevated temperatures required for thermal condensation between
carboxylic acids and amide. They may also be too sensitive chemically to the
conditions necessary for the formation of an acid chloride (see below).
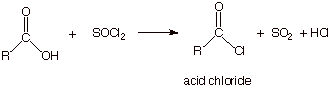
o
A chemically
and thermally mild method
for amide formation would therefore be extremely useful. Such a method is the
room temperature condensation between carboxylic acids and amines catalyzed by
dicyclohexylcarbodimide (R’’’ = cyclohexyl).
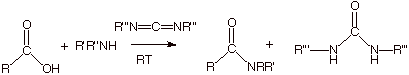
o
This method
is extensively used to prepare peptides from amino acids.
o
Incidentally,
the diimide is neither strongly basic nor acidic. It is, however, basic enough
to be protonated on one of the imide nitrogens by the carboxylic acid. We will
not write out the detailed mechanism for this powerful method for the catalysis
of amidification. However, please note that the water produced in the formation
of the amide link is effectively removed by combination with the diimide to
yield a derivative of the very thermodynamically stable molecule urea.
NITRILES.
q
Nitriles are
organic cyanides. They are often prepared by SN2 reactions of the
powerfully nucleophilic cyanide anion with an organic halide.
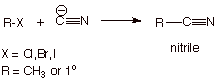
q
Note that
the nitrile carbon is in the +3 oxidation state, so that although it is not an
acyl derivative, it is at the same oxidation level as the other acyl
derivatives.
q
Therefore,
nitriles can be hydrolyzed to carboxylic acids or esters by either acidic or
basic hydrolysis.