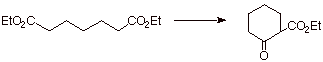Chapter 19: Enols and Enolates
of Carbonyl Compounds and Their Reactions
We have seen that
the carbonyl group of aldehydes and ketones is highly reactive, and that additions to this functionality are common. In the present
chapter we will see that not only is the carbonyl functionality reactive per
se, but that it also activates nearby
carbon-hydrogen bonds (specifically alpha hydrogens) to undergo a variety of substitution reactions.
ENOLS.
q
Enols are isomers of
aldehydes or ketones in which one alpha hydrogen has been removed and replaced
on the oxygen atom of the carbonyl group. The resulting molecule has both a C=C
(-ene) and an –OH (-ol) group, so it is referred to as an enol. Strictly speaking, to be an enol the –OH and
the C=C must be directly attached to one another, i.e., in conjugation with
each other, as shown below.
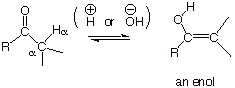
q
We shall see that enols
can be formed either by acid or base catalysis and that, once formed, they are
highly reactive toward electrophiles (i.e., they are pretty strong nucleophiles).
We shall also see that not all carbonyl compounds can form enols, but only
those which have hydrogens of the alpha type. The carbon of an aldehyde or the
two carbons of a ketone which are directly attached to the carbonyl carbon are
designated as alpha carbons, and any hydrogens directly attached to these carbon atoms are termed alpha
hydrogens. There can be more than one type of alpha hydrogen, or there may be
no alpha hydrogens in a given carbonyl compound.
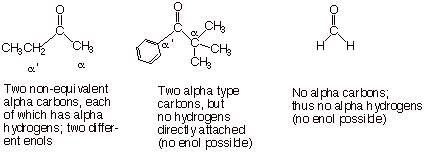
q
We should not be
surprised at all that in the
equilibrium between a carbonyl compound and its corresponding enol, the
equilibrium lies well to the carbonyl side, i.e., usually only small amounts of
enol are present in the equilibrium. The preference for the carbonyl form over
the enol form derives from the well-known circumstance that the C=O function is
imuch more stable than the C=C function.
Mechanism of Acid-Catalyzed Enolization
q
Enol formation is called
“enolization”. The mechanism whereby enols are formed in acidic
solution is a simple, two step process, as indicated below:

q
Step 1 is simply the
protonation of the carbonyl oxygen to form the conjugate acid of the carbonyl
compound. Remember that proton transfers from oxygen to oxygen are virtually
always extremely fast. The equilibrium of the first step is established very
quickly.
q
The second step is the
removal of an alpha proton from the conjugate acid by water acting as the base.
Although both steps are relatively fast, this is the slower of the two steps. The transfer of a proton from
carbon is not as fast as that from oxygen, in general.
q
The overall result is
addition of a proton to the carbonyl oxygen and removal of a proton from the
alpha carbon, in that order.
q
We should also note that
the reverse of the second step, the protonation of the enol form, occurs at the carbon which we call the
beta carbon of the enol (the same carbon which was called the alpha carbon in
the context of carbonyl chemistry) to go back to the same conjugate acid as was
formed by the protonation of the carbonyl form. A key point here is that this conjugate acid is the common
conjugate acid of both the carbonyl
compound and its enol form. It is the intermediate which allows them to be
rapidly equilibrated. By considering the resonance structures for this
conjugate acid, shown below, you can see how they relate in structure to both
the carbonyl form (the oxonium structure) and the enol form (the carbocation
structure).

q
When this conjugate acid
loses a proton from the oxygen atom, it goes to the carbonyl compound. When it
loses a proton from the alpha carbon, it goes to the enol form. Depending on
whether the proton is lost from oxygen or carbon, this conjugate acid can
deliver either the carbonyl form or the enol form.
Structure and
Resonance Stabilization of the Enol.
q
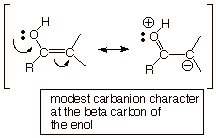
Since the unshared pair
of electrons on the hydroxyl
oxygen and the pi electrons of the alkene double bond are directly connected so
as to be in conjugation, the two groups interact, resulting in delocalization
of the electrons and resonance stabilization.
q
Note that the second
resonance structure has charge separation, so it is not as low in energy as the
first. As a result, the resonance stabilization is only modest..
q
The pi bond of an alkene
is already nucleophilic, but that of an enol becomes even more electron-rich
because of the carbanion character generated at the beta carbon. Enols
therefore react extremely rapidly with electrophiles. We have already discussed
their protonation. We will see that other electrophiles like bromine also react
rapidly and at the beta carbon in particular.
Relative Stabilities of the Carbonyl and Enol Isomers
In a typical
carbonyl/enol equilibrium, the equilibrium constant is about 10-5 ,
i.e., there is only about .001% of the enol. As briefly mentioned previously,
this reflects the greater stability of the C=O double bond than the C=C double
bond.
Mechanism for Base-Catalyzed Enolization
q
As noted before, the
enol can be generated by either an acid- or a base-catalyzed mechanism. Incidentally,
at pH 7 enolization is very slow, so that either acid or base is required for
enolization.

q
As in acid-catalyzed
enolization, the slower step is the removal of the alpha proton.
q
It is important to note
that the enolate is the conjugate base of both the carbonyl compound and the
enol form. If the enolate is protonated on oxygen, it generates the enol (step
2 of the above mechanism), but if it protonates on carbon (see the reverse of
step 1), it generates the carbonyl compound. Since there is partial negative
charge on both of these, protonation can occur readily on either one.
q
Also important is the
fact that this alpha proton is acidic enough to be removed rapidly by the
strong base, hydroxide ion. The pKa of a carbonyl compound which has
an alpha hydrogen is typically about 19, only about three powers of ten less
than the pKa of water (17.7). Note that the equilibrium lies on the
side of the weaker acid (the carbonyl compound; water being the stronger acid),
but the K value is ca. 10-3.
q
Why is this proton so
acidic, when C-H bond protons of alkanes typically have pKa’s
of 50? Recall that anion stability
is usually the controlling factor in determining relative acidities. The anion
formed in this case is an enolate anion which is highly resonance stabilized,
as shown above.
q
Recall that the rules of
resonance specify that large resonance stabilizations result when there are 2
or more structures of equal energy.
Usually, this happens when, as in the case of carboxylate anions, the two
structures are equivalent. However, equal or near equal energies can result
even when two canonical structures are not symmetry-equivalent. This is the
case with an enolate anion.
q
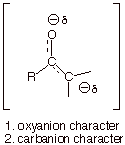
In one structure, there
is carbanion character and in the
other oxyanion character. The
latter, is of course, substantially more favorable energetically. However, the
structure which has carbanion character has a carbonyl group, which is more
favorable than the alkene double bond present in structure having oxyanion
character. Each structure has one favorable and one unfavorable character,
leading to an approximate equality in energy for the two structures. Consequently, the resonance
energy is relatively large.
Relative
Amounts of the Enolate, Enol, and Carbonyl Compound.
q
We have already seen
that the enol is a minor (but mechanistically important) component of the
carbonyl/enol equilibrium. The position of this equilibrium is, of course,
unaffected by the pH, since it depends only upon the relative free energies of
the carbonyl and enol components. On the other hand, since the pKa
of carbonyl compounds having alpha type hydrogens is only ca. 10-19,
there is a negligible amount of enolate present in aqueous solution at neutral
pH:
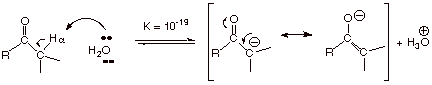
q However, the base hydroxide ion is much more powerful than the base water, so that the equilibrium --- in this case---is much more favorable, although still lying somewhat on the carbonyl side.
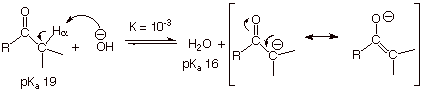
q
However, it will be
useful for us to know that a still stronger base than hydroxide ion, e.g., the
amide ion, can quantitatively convert the carbonyl compound to its enolate.
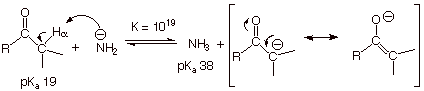
q
In contrast, at acidic
pH’s, the enolate concentration is too miniscule to warrant
consideration.
q
As a result of these
considerations we can see that at neutral or acidic pH’s, there is more
enol than enolate. For example, at neutral pH, the K for enolization (10-5)
is much larger than the K for acid dissociation (10-19). But in the
presence of hydroxide anion, there is more enolate than enol, because the
equilibrium constant for conversion of an carbonyl compound to its
corresponding enolate (10-3) is larger than the K for enolization
(10-5).
Acid-Catalyzed Bromination
We have seen that
the enol can be generated by acid or base-catalyzed mechanisms. Also, we have
seen that the enol contains an
electron-rich alkene functionality, which should be highly reactive
toward electrophiles. Bromine is a very reactive electrophile, even toward
simple alkenes. It should be and is enormously reactive toward enols. The
net result is the substitution of a bromine atom for one of the alpha hydrogens
of the carbonyl compound. Note also that carbonyl compounds without alpha
hydrogens do not react with bromine at all.
Mechanism of Acid-Catalyzed Bromination
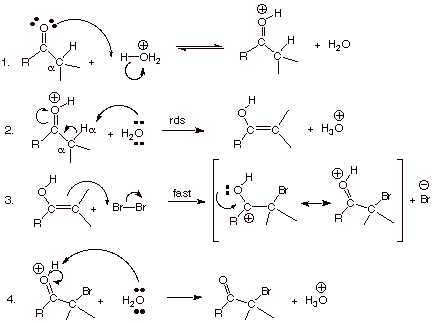
q
The first two steps are
essentially identical to those in acid-catalyzed enolization. It is the enol,
not the carbonyl compound, which is reactive toward bromine. The sole
difference is that in the case of bromination, the second step is actually rate-determining
(rather than just “slow”).
This is because the enol is so reactive toward bromine that it never has a
chance to reverse step 2, i.e., it is not protonated by hydronium ion to give
back the conjugate acid of the carbonyl compound. So bromine reacts much more
rapidly than does hydronium ion with the intermediate enol. Once formed, the
enol always goes on to brominated product. The rate of enol formation is
exactly equal to the rate of formation of the brominated product.
q
An interesting and important consequence
of the fact that bromine does not take part in the reaction until after the rds is that the reaction rate (the rate of
consumption of the carbonyl compound or the rate of formation of the brominated
carbonyl compound) is independent of the concentration of bromine. If we double the concentration of bromine, the rate
remains exactly the same or if we cut the concentration in half, the rate is
not diminished at all. This is because the formation of the enol is
rate-determining.
q
Even further, if we use
chlorine rather than bromine, the rate is still the same as for bromination. The
rate is not only independent of the concentration of the halogen but also of
the nature of the halogen. Even though bromine is more reactive than chlorine,
in general, in electrophilic additions, chlorination and bromination both occur
at exactly the same rate.
Base-Promoted Bromination
As we have noted,
when hydroxide ion is present, both the enol and enolate are typically present
in equilibrium with the carbonyl component. We have also seen that under these
basic conditions the enolate is the predominant form. However, the enolate is also the more reactive
form toward electrophiles. For both
of these reasons the enolate is the reactive species of interest in basic
solutions. Please keep in mind also that the carbonyl component, which is
present in great excess over both the enol and enolate, is essentially
unreactive toward bromine and many other electrophiles ( excluding, of course,
hydronium ion).
Mechanism:
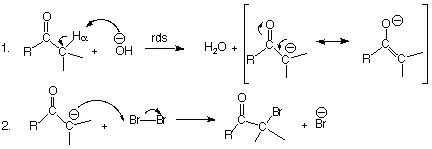
q
Again, as with
acid-catalyzed bromination, bromine does not appear in the reaction until after
the rds. The enolate reacts much more rapidly with bromine (step 2) than with
water (reverse of step 1). So step 1 is never reversed, and the enolate once
formed always goes on and goes on rapidly to the product.
q
Note that the canonical
structure of the enolate which gives it carbanion character is not a
charge-separated structure, as it is in the case of the enol. So the enolate
has much more carbanion character than the enol, and is much more reactive
towards electrophiles.
THE ALDOL REACTION
We have seen that
the enol form of a carbonyl compound, though only a minor constituent in the
equilibrium mixture, is vitally important in the reaction with electrophiles.
The same is true of the enolate in basic solutions. We have also seen that the
enolate is a more potent nucleophile than the enol. Since the carbonyl group of
the carbonyl form is strongly electrophilic and reacts with a variety of
nucleophiles, it would be reasonable to expect the strongly nucleophilic
enolate to be able to add to the carbonyl group of the carbonyl component
(which is the major component in the equilibrium).
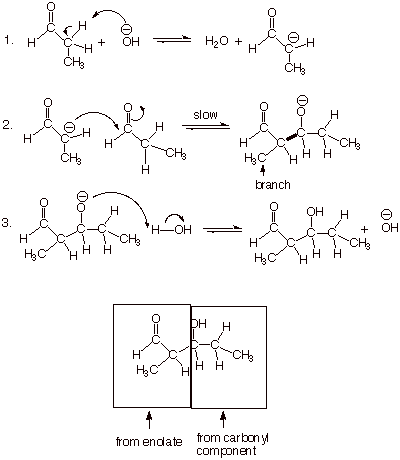
Mechanism of the
Aldol Addition Reaction.
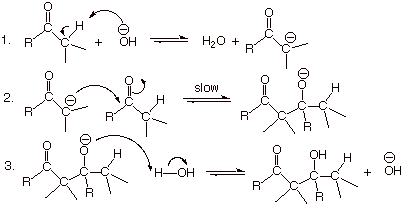
q
The “slow”
step is the addition of the highly nucleophilic enolate to the electrophilic
carbonyl carbon of the relatively strong carbonyl pi bond. As with most
carbonyl additions, this reaction is reversible, so it is not rate determining.
q
Note that the aldol
product is so-called because it has both an aldehyde and an alcohol moiety. It
is specifically called an aldol
addition because it works better with aldehydes than ketones (recall that the
carbonyl pi bond of ketones is thermodynamically more stable than that of
aldehydes).
q
The special
importance of the reaction, is that it can be used to construct new
carbon-carbon bonds, the most essential aspect of organic synthesis.
q
Since carbonyl compounds
which do not have alpha hydrogens can not form an enolate, they cannot undergo
the aldol reaction. Therefore the simplest aldehyde, methanal (formaldehyde)
cannot undergo the aldol reaction.
The simplest
case: Aldol Addition of Ethanal (Acetaldehyde)
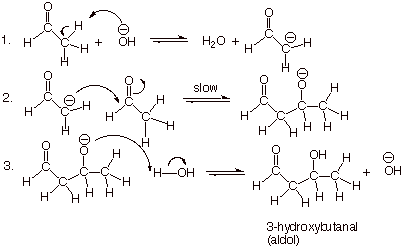
q
Note that the hydroxyl
group is always formed at the 3-position of the aldol product.
q
Also, please remember
that there are two distinct “roles” in the aldol reaction for the
aldehyde. These are: (1) the enolate role and (2) the carbonyl role. In viewing
a given aldol product, you can see which portion of the product arose from the
enolate and which from the carbonyl component.
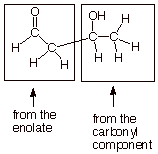
Predicting the Structure of Aldol Addition
Products
q
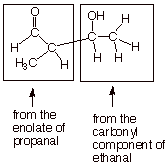
When an aldehyde more
complex than ethanal is used, the structure of the aldol addition product is
slightly more difficult to predict, since it will typically have one or more branches in the chain. Consider the product of the aldol
reaction of propanal.
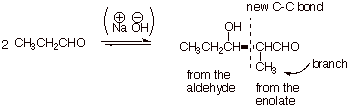
q
The branch,
incidentally, always occurs on the enolate side of the aldol product. It is
there because only the alpha hydrogen can be removed to form the enolate,
leaving any other more remote carbons of the aldehyde as a branch.
q
You should be able to
:(1) Write the detailed mechanism for the aldol addition reaction of any
specified aldehyde (2) Draw the
structure of the appropriate aldol product from any specified aldehyde without
having to write out the mechanism (3) Indicate, for any aldol product, which
part is derived from the enolate and which from the carbonyl component .
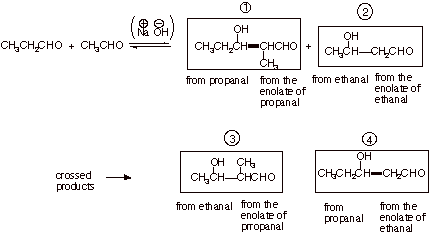
The Crossed Aldol
q
It has been noted that
in the aldol addition reaction, there are two distinct roles, that of the enolate
and that of the carbonyl compound. But in this reaction, a single carbonyl
compound is used as the source of both the enolate and the carbonyl components.
Consequently many structures which might be desirable are not accessible
because they would require two different components. Consider the
hydroxyaldehyde given below:
q
If one attempted an
aldol reaction between ethanal and propanal, this aldol would indeed be one of
the products, but it would only be one of four different products (there are two
enolates and two carbonyl components, resulting in four possible combinations,
all of which are realized. Consequently, the crossed aldol is not a generally
feasible reaction.
q
However, it has been
noted that a number of carbonyl compounds, those which lack alpha hydrogens,
are not able to form enolates at all. In many cases it is possible to perform
crossed aldol reactions between one of these non-enolizable carbonyl compounds
and a general aldehyde. In the examples below, benzaldehyde, pivaldehyde, and
methanal are the non-enolizable carbonyl compounds.
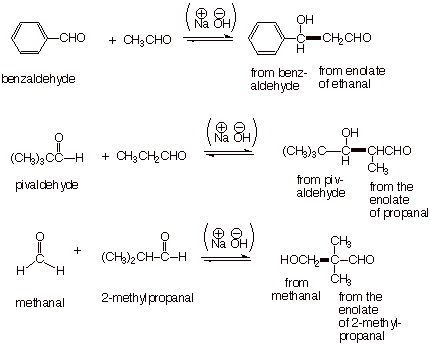
q
You may notice that
there are still two possible aldols since, although there is only one enolate,
there are still two possible carbonyl components. That is, we can get the
crossed aldol shown, but we can also get the normal aldol of the enolizable
component.
q
In order to minimize the
formation of the normal aldol product and maximize the yield of the crossed
aldol product, the non-enolizable carbonyl component is kept in excess by
adding the enolizable componet in a dropwise fashion to the basic solution of
the non-enolizable component. Thus, when the enolate is formed, it always has a
greater opportunity to react with the non-enolizable carbonyl component.
The Aldol Condensation Reaction
A condensation
reaction is one in which water or
another small molecule, such as methanol, is formed in a reaction between two
organic molecules. The aldol condensation reaction is a reaction which starts
just like the aldol addition, but then subsequently the aldol adduct undergoes
a further reaction, the elimination of water to generate a C=C bond in place of
the alcohol function. The simplest aldol condensation reaction is illustrated
below:
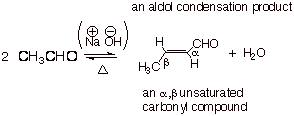
q
In an overall sense, the
carbonyl oxygen of one molecule and two alpha hydrogens of another are
eliminated as water. The reaction proceeds first to give the aldol addition
product, by exactly the same mechanism as indicated previously, but because of
the higher temperature employed,
this aldol adduct undergoes dehydration to yield the aldol condensation
product.
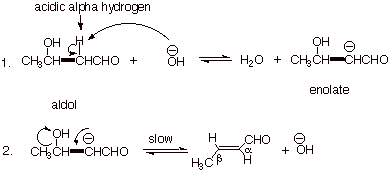
q
The formation of the
enolate in base is expected, but the elimination of hydroxide anion is,
perhaps, a little surprising, and indeed it is the slow step. The mild surprise
might be that hydroxide ion is considered to be a rather poor leaving group,
and it is. However, in the context of an intramolecular reaction in which the
negative charge is in the same molecule as the hydroxyl group, it often occurs.
The Claisen Ester Condensation
The carbonyl group of
an ester functionality also activates any alpha hydrogens which may be present.
But, since the ester group has additional resonance stabilization beyond that
of a carbonyl (ester resonance), it does not activate these hydrogens quite as
much as does an aldehyde or ketone. This can be seen in the pKa
values of esters which have alpha hydrogens (ca. 21). Nevertheless, the enolate
of an ester can be formed in small but sufficient quantities for the purposes
of many reactions simply by treating the ester with a strong base such as
ethoxide anion.
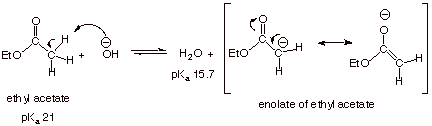
q
In a manner very similar
to the aldol condensation of aldehydes and ketones, the enolate of an ester can
add to the carbonyl group of another ester molecule. The overall result of this
reaction, which is called the Claisen ester condensation, is the formation of a
beta ketoester (as compared to a beta
hydroxyester in the aldol addition
reaction).
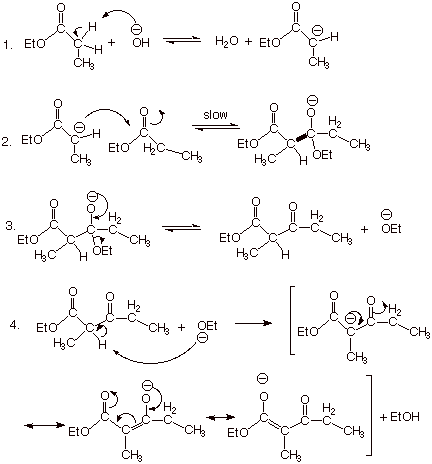
q
There are several new
features to this mechanism, but the first two steps are essentially the same as
for the aldol reaction. In the third step, however, an ethoxide anion is
eliminated, resulting in the formation of a ketone function. Essentially, this
step is the reverse of hemiacetal formation from a ketone function and ethoxide
anion, and we know that this equilibrium is favorable to the ketone side of the
equation.
q
Another new, and
exceedingly important, feature of
this mechanism is found in the fourth and final step. An alpha type proton is
abstracted by the ethoxide ion in a reaction which (please note) goes to completion. This particular hydrogen is alpha to two carbonyl
groups, an ester carbonyl and a ketone carbonyl. As such it is especially
highly acidic, so that the equilibrium goes to completion.
q
The pKa’s
of protons which are alpha to two carbonyl groups is typically ca. 10. Since
the acid on the right hand side of
equation 4 (ethanol) has a pKa of 16, this latter acid is the
weaker acid, and the equilibrium proceeds far to the right. In fact the K value
is ca. 106 (remember
how to calculate K’s from the Ka’s of the reactant and
product acids?).
q
The reason for the
especially high acidity of the beta keto ester, as we have often seen, lies in anion
stability. The conjugate base of this
substrate is highly resonance stabilized, having three relatively good resonance structures (recall that a
ketone enolate only has two).
q
It is important to note
that without this highly favorable (exergonic) neutralization reaction, the
Claisen ester condensation would not work at all. This reason for this is as
follows: The net result of steps 1-3 of the mechanism is the conversion of one
ester function to a ketone function, thus forfeiting the substantial ester
resonance. However, the final exergonic step drives the reaction to completion.
q
As we have seen in other
instances before, this means that the reaction is not catalytic, but promoted,
which means that we have to use a stoichiometric amount of ethoxide anion, in contrast to the aldol condensation, which
is catalytic in base. Finally, To obtain the ultimate product, the beta keto
ester, we have to acidify the solution with strong acid in the aqueous workup.
[Why wouldn’t just a neutral aqueous workup do the job?].
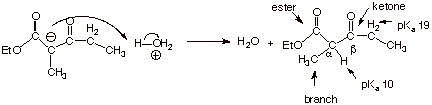
The Dieckmann Condensation: An
Intramolecular Version of the Claisen Ester Condensation.
When a difunctional
ester is used, if the ester groups are positioned so that there are 4 or 5
carbon atoms between them, an intramolecular version of the Claisen
condensation can occur and is especially useful in the construction of five and
six-membered rings.
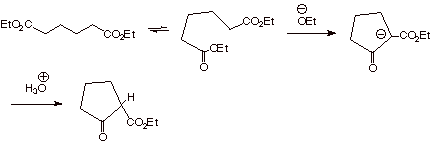
q
Although the extended
(all anti) conformation of such a
diester is the conformational minimum, other conformations are possible, and
are substantially populated by facile rotations around the carbon-carbon single
bonds, in which the alpha carbon of one ester function is very proximate to the
carbonyl group of the other ester function (as shown in the first equilibrium
above). This can lead to a cyclization reaction between the ester enolate and
the other ester function present in the same molecule, affording a 5 or 6
membered ring.
q
The mechanism of the
reaction is the same as for the standard Claisen condensation and is shown
below:
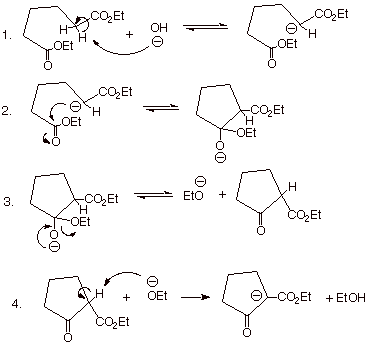
q
This works efficiently
because the enolate, once formed, reacts more rapidly with the carbethoxy group
in the same molecule (i.e., intramolecularly) than with a carbethoxy group of
another molecule (intermolecularly).
q
In effect, when the
second ester function is present in the same molecule as the enolate, the
effective concentration of the ester function is much higher than in the bulk
solution.
q
This kind of reaction is
an excellent way to synthesize relatively unstrained five and six membered
rings. The following ester would yield a six-membered ring.