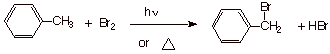Chapter 20: Benzene and Derivatives:
Aromaticity
Recall that
resonance stabilization is especially strong when structures of equal energy
are available, as in the case of the carboxylate anions. However, resonance
stabilization rises to its highest level when not only are equivalent
structures available, but the conjugated system is cyclic and has 4n+2 pi electrons in the cyclic system. Such cyclic, conjugated systems
are sometimes referred to as aromatic.
q
The 4n+2 Rule is also sometimes called the Huckel Rule. Recall that n can be 0,1,2,3---, so that the systems
which are highly stabilized have 2,6,10,14,- pi electrons in a cyclic conjugated system.
q
By the way, the 4n+2
Rule is not applicable at all to acyclic conjugated systems, where 4n+2
electron systems are no more or less stable than 4n conjugated systems.
q
We will also see that
when a cyclic, conjugated system has 4n pi electrons (4,8,12,--electrons), the
system is not especially stabilized and can be unusually unstable. Such systems
are called antiaromatic.
Benzene
As you may recall,
benzene is the prototype aromatic
system. Originally, the chemical term “aromatic” referred to the
aroma of benzene and other derivatives of benzene. In the modern context, it
refers to the unusually great resonance stabilization of benzene and closely
related derivatives of benzene and the common reactivity patterns they exhibit
as a result of this extraordinary stabilization.
q
Benzene, of course, has two
equivalent resonance structures,
which are called Kekule structures.
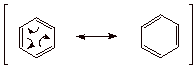
q
It has 6 pi electrons in a single cyclic conjugated system. You can count the number of electrons in the system
by examining any canonical structure and counting two electrons per pi bond.
q
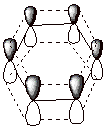
An orbital picture (a representation of the constituent 2p AO’s
which overlap to form the conjugated pi electron system) is presented below.
Each carbon is sp2
hybridized, having C-C sigma bonds to two other carbons along with one C-H
sigma bond. All three of these sigma bonds lie in the common trigonal plane and
have form ca. 120 o bond angles with each other. Therefore, each of
the six carbons has one remaining 2pz AO which is perpendicular to
the trigonal plane. These six 2p AO’s overlap in a cyclic fashion to form
a delocalized conjugated system, i.e., all of the resulting MO’s are
delocalized over the whole system of six carbon atoms.
q
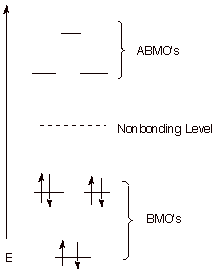
An Energy Level Diagram
for the MO’s of benzene is depicted below. In examining this diagram,
recall that cyclic overlap between the six AO’s converts them to six delocalized
MO’s, and that of these six
MO’s, three are BMO’s
and 3 ABMO’s. It is
especially important to note, in terms of the 4n+2 Rule, that three bonding
molecular orbitals can just accommodate six electrons, the correct number for
aromatic stabilization. Any additional electrons would have to go into high
energy, antibonding MO’s, which would detract from the stabilization of
the system. Any fewer electrons would leave one or more BMO’s unfilled,
thus providing less than the maximum stabilization.
q
In regard to the display
of MO’s in the energy level diagram, please recall that typically (the
exceptions we will soon see), the MO’s are symmetrically distributed
about the nonbonding level (the
dashed line, which represents the energy of a carbon 2pz atomic
orbital). For example, there is a degenerate pair of BMO’s and a
corresponding pair of NBMO’s essentially equidistant in energy terms
below and above the NBMO level.
q
Also there is, in such
cyclic systems, always a unique (non-degenerate), lowest energy BMO. The rest
of the MO’s occur in degenerate pairs up to the highest energy ABMO which
is symmetrically related to the unique bonding MO.
q
As a consequence, there
is always an odd number of BMO’s in aromatic systems, i.e., 1,3,5,7. This means that when all of the
BMO’s are filled, there are 2,6,10,14, etc. electrons.
Definition of Aromaticity. Powerful resonance stabilization, similar to that of
benzene, which is found in cyclic conjugated pi electron systems having 4n+2 pi
electrons.
Reactivity consequence. Benzene, unlike alkene and carbonyl pi systems, does
not typically undergo addition reactions, which would disrupt this exceptional
resonance stabilization. Instead, benzene tends to undergo substitution
reactions which preserve the aromaticity.
Magnitude of the Resonance
Stabilization. Estimates of the
resonance stabilization of benzene vary, but it is generally considered to have
no less than 36 kcal/mol of such stabilization.
Aromaticity in Cations and Anions.
Aromaticity depends
upon the number of electrons in the cyclic conjugated system (the electron
count), and not upon either the size
of the ring or whether it is neutral or negatively or positively charged. As a
consequence, there are quite a number of aromatic anions and cations.
An Aromatic Anion. The rather simple hydrocarbon 1,3-cyclopentadiene, which contains no electronegative atom to stabilize the negative charge in its anionic conjugate base, is essentially as acidic as water, where the conjugate base (hydroxide anion) is stabilized by the highly electronegative oxygen atom. The amazing acidity of this hydrocarbon is of course the result of anion stability, in particular the aromatic resonance stabilization of the anion.
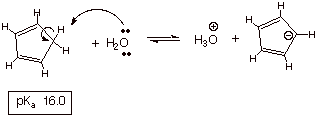
q
Note that the anion has
a cyclic conjugated system of pi electrons (each carbon atom is sp2 hybridized and has a 2pz orbital on it. The
electron count is 6, just as in the
case of benzene, satisfying the 4n+2 rule for aromaticity of cyclic conjugated
systems.
q
The electron count is
obtained by examining any one canonical structure of the anion, and counting 2
electrons per pi bond and 2
electrons for a carbanion center (the
carbanion center has a 2p AO which is doubly occupied).
q
In terms of resonance
theory, the cyclopentadienide anion has five equivalent resonance structures, distributing the negative charge equally over all
five carbon atoms. The anion therefore has five-fold symmetry.
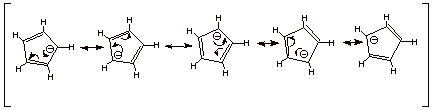
q
The acidity of
1,3-cyclopentadiene is so great (for a hydrocarbon), that this anion can be
generated in substantial amounts (but
not quantitatively in the sense that the reaction goes to completion) in
basic aqueous or alcohol solution
(recall that the pKa is essentially the same as that of water or
methanol or ethanol, so that the equilibrium constant is ca. 1.
q
The MO Picture. As was seen in the case of benzene, there is a
unique, highly bonding BMO, followed by degenerate pairs of MO’s. Since
there are only five atoms in the system (5 AO’s from which to build
MO’s), there are only 5 MO’s. As is evident from the MO energy
level diagram below, the symmetrical relationship of the BMO’s vs the
ABMO’s about the non-bonding level is lost in systems containing an odd
number of atoms in the conjugated system.
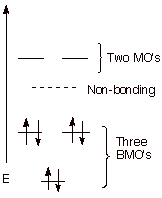
q
Again, there are three
BMO’s, so that six electrons is the perfect number for gaining the
maximum stabilization.
q
The Circle
Mnemonic. The display of the MO’s
for a given cyclic, conjugated system can be obtained by means of a simple
mnemonic (memory) device. A polygon of the appropriate size is inscribed inside
a circle, with one vertex pointing downward. The vertices of the polygon then
correspond to energy levels on a vertical energy scale. In the case of the
cyclopentadienyl system, a pentagon is inscribed inside a circle. For benzene,
a hexagon, etc.
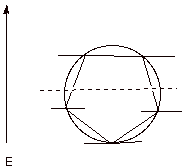
Aromatic Carbocations. Carbocations, species which contain trivalent, positively charged carbon, are familiar intermediates, but they are typically highly reactive, short-lived intermediates. This is true even for a relatively stabilized carbocation like the tert-butyl carbocation. However, if the carbocation moiety is contained in a cyclic, conjugated system having 4n+2 pi electrons, the carbocation may be stable enough even to isolate as a salt.
q
A particularly
impressive example of an aromatic carbocation is the cycloheptarienyl
(tropylium) carbocation,. This cation, like benzene and the cyclopentadienyl
anion, has a six pi electron aromatic ystem. Again, the electron count is
properly obtained by counting 2 electrons per pi bond and zero electrons for a
carbocation center (vacant 2p AO) in any canonical structure.
![]()
q
This cation can be
isolated as a salt with various counteranions, including the tetrafluoroborate
anion. It is stable in aqueous solution.
q
An isolable aromatic
carbocation can be prepared in the highly strained cyclopropenyl system. Note
that in the case of a cyclopropenyl system there is only one BMO, so that the
aromatic system contains 2 electrons (4n+2, with n = 0).
![]()
q
The circle mnemonic and
the resulting display of MO’s for the cyclopropenyl system is illustrated
below:
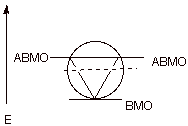
q
Stable aromatic anions
and cations having 10, 14, and many higher electron count systems have also
been prepared.
Heterocyclic
Aromatics. The aromatic systems which we
have previously considered were all carbocyclic, i.e., they contained only
carbon atoms in the cyclic conjugated systems. Other non-carbon atoms (called
“heteroatoms”) can also form a part of aromatic systems, giving
rise to a wide variety of such systems. Of these heteroatoms, oxygen and
nitrogen are perhaps the most common.
q
Recall that the oxygen
atom of water or an alcohol or an ether is sp3 hybridized and has
two unshared electron pairs in two of the four tetrahedral orbitals. However,
we have also seen that the oxygen atom of an ester which is singly bonded to
the carbonyl carbon is sp2 hybridized. This was also seen to be
true, incidentally, for the nitrogen atom of an amide). Why is this? Simply
because this places one of the oxygen electron pairs in a 2pz AO,
and we know that the latter type of AO is optimum for the pi overlap that gives
rise to ester type resonance. Note that the other unshared pair is in an sp2
AO in the trigonal plane of the oxygen and the carbonyl group and is not a part
of the ester conjugated system.
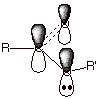
q
Oxygen as a
Heteroatom in Aromatic Systems. In the
same way, an oxygen atom placed in a cyclic, conjugated system prefers to be sp2
hybridized in order to maximum the cyclic conjugation and resonance
stabilization. Furan is a simple example of such a heterocyclic aromatic
containing oxygen. Structure 1
below gives a typical structure for furan, indicating the two unshared pairs.
Structure 2 shows the unshared
pair in the 2pz AO, and structure 3 shows both the latter unshared pair and the one in an
sp2 AO which is not part of the conjugated system. The electron
count therefore is six. Two from each of the two pi bonds and two from the
oxygen 2pz AO. The two electrons in the sp2 AO are not
counted.
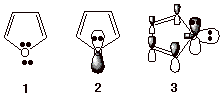
q
We would also like to
note that conjugated system of furan is closely analogous to that of the
cyclopentadienyl anion, both having five atoms and six electrons in the
conjugated system. Furan, however, is electrically neutral, while the
cyclopentadienyl anion has a unit of negative charge. We have previously
asserted that the existence or sign of charge has little effect upon
aromaticity.
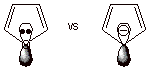
q
Nitrogen as
a Heteroatom in Aromatic Systems. The Pyrrole Model. Unlike oxygen, nitrogen has two ways in which it can
be incorporated into aromatic systems. The first mode of nitrogen incorporation
is illustrated by pyrrole, an aromatic compound analogous to furan.
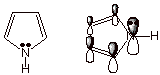
q
Unlike oxygen, nitrogen
is trivalent and has only one unshared electron pair. This occupies a 2pz
AO and becomes a part of the conjugated system, as in the case of furan.
Instead of the other unshared pair that oxygen has in its trigonal plane,
nitrogen has another valence. In the simple case of pyrrole, this other valence
is an N-H bond, but many other atoms or groups could be bonded to nitrogen and
still preserve the aromaticity.
q
Nitrogen as
a Heteroatom in Aromatic Systems. The Pyridine Model. Since nitrogen is trivalent, it can also
be doubly bonded to carbon, i.e., it can form a C=N bond in the context of an
aromatic system. In this case, nitrogen is using its only 2pz AO to
form a double bond in the conjugated system, so that the unshared pair is in an
sp2 AO in the trigonal plane. Consequently, the latter pair is not
counted as part of the aromatic system. Nitrogen, then, only contributes one
electron to the conjugated system, as does any of the carbon atoms of benzene,
e.g. Pyridine, a heteroatomic model of benzene, is the prototype example of
this kind of heteroaromaticity.
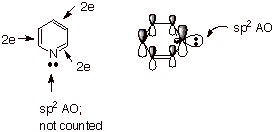
q
By way of distinction
between the two different modes of nitrogen participation in heteroaromaticity,
if the nitrogen atom has a valence extended outside of the ring, as in the case
of pyrrole (the N-H bond), it contributes two electrons to the system. In this
case, nitrogen behaves like oxygen. If it has no valencies outside of the ring,
it contributes one electron to the aromatic system. Its behavior in this
situation is like that of carbon (e.g. in benzene).
Antiaromatic
Systems. Cyclic, conjugated systems
having 4n pi electrons, instead of
the “magic number” of 4n+2 pi electrons are not aromatically
stabilized, and in some instances are especially unstable systems. These are
sometimes designated as antiaromatic systems.
Highly Unstable (Antiaromatic) Systems. 1,3-Cyclobutadiene, the cyclopropenyl anion, and the cyclopentadienyl cation are prime examples of cyclic conjugation which results in a surprisingly unstable system.
q
Cyclobutadiene has four
pi electrons. The MO display (recall
the circle mnemonic) is illustrated below, showing that the second pair of
electrons must go into a nonbonding orbital. (or if the Hund Rule is followed, one electron into
each of the two NBMO’s). Thus, this second electron pair produces no
bonding at all. In effect, we have
something like the equivalent of just one double bond, although the canonical
structure indicates that there are two.
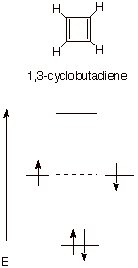
q
Correspondingly,
cyclobutadiene is only stable enough to be examined spectroscopically at
temperatures near O K. At 30K, it already decomposes rapidly!
q
An interesting and
paradoxical situation arises in regard to the resonance theoretical picture of
cyclobutadiene. As in the case of benzene, there are two equivalent
resonance structures. This would seem
to suggest that the two systems might have comparable resonance stabilizations.
Instead, benzene is highly stabilized, and cyclobutadiene is actually
destabilized. What is the problem? The problem is with the resonance approach,
which does not understand, as the MO method does, that there is a magic number
of electrons. Consequently, the resonance method is not reliable in the case of
antiaromatic systems.
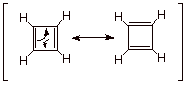
q
The other 4n systems
mentioned above which are also often classified as antiaromatic are the
cycloproenyl anion and the cyclopentadienyl cation.
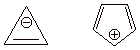
q
Other 4n pi electron
cyclic conjugated systems which have 8,12, etc. electron counts are usually
considered to be simply non-aromatic,
i.e., they do not appear to be highly stabilized or destabilized. An example of
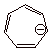
an 8 pi electron non-aromatic system is the cycloheptatrienyl anion. This anion
is moderately stable, but nowhere near as stable as the cyclopentadienyl anion.
Polybenzenoid
Aromatics.
There is an
extensive series of polycyclic aromatic systems which are based upon the
benzene structure. Essentially, these systems have two or more benzene rings fused
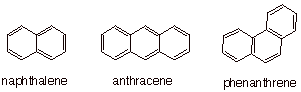
to each other via a common C=C bond. Naphthalene is the simplest of this series, having two fused
benzene rings. Although naphthalene has 10 pi electrons and its aromaticity
could be considered to be based upon having the magic number of electrons, it
should be noted that the 4n+2 rule does not strictly apply to polycyclic
systems. The existence of two such tribenzenoid aromatics, anthracene and phenanthrene illustrates the possibility that benzene rings can be fused in a linear (anthracene) or an angular (phenanthrene) fashion. Many higher polycyclic
aromatics are know, and some of them are powerful carcinogens (PBA’s are polybenzenoid aromatics).
Arenes.
Arenes, or
substituted benzenes, are derivatives of benzene in which one or more (up to all six) of the six
hydrogens of benzene is replaced by another substituent or substituents. Of
course, naphthalene and polybenzenoid aromatics also have analogous
derivatives. Some common examples
of monosubstituted arenes are shown below. Note that many of them have
non-systematic, but IUPAC-approved names (toluene, phenol, aniline).
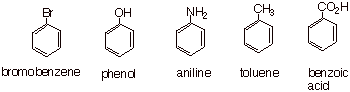
Phenols.
Phenol is essentially hydroxybenzene. Since it has an –OH functional group, it is an
alcohol. However, we have previously seen that when an –OH group is
directly attached to a carbonyl function, the result is a compound
functionality, the carboxyl group, which has distinctly different behavior from
typical alcohols. When an –OH group is directly attached to a benzene
ring, a similar but somewhat less dramatic change in properties occurs. Consequently,
phenols (or any species in which the –OH group is attached to any
aromatic ring) are considered to be a special sub-class of alcohols, with
significant differences, but also many parallels, in their behavior in
comparison to ordinary alcohols.
q
Acidity: the acidity of phenol (pKa 10) is about a
millionfold greater than that of simple alcohols, which have pKa’s
of ca. 16.
![]()
q
This strongly enhanced
acidity is the consequence, in general terms, of anion stability, and in specific terms of the extensive resonance
stabilization of the conjugate base of phenol, the phenoxide ion.
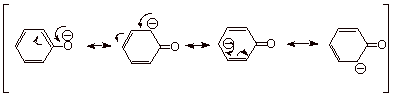
q
These structures
indicate that the negative charge resides partly on the phenoxide oxygen, but
also is delocalized in part onto the benzene ring. In particular, partial negative
charge appears on the 2,4, and 6 positions of the benzene ring. The 2 and 6
positions are equivalent and are referred to as ortho positions, while the 4 position is referred to as the para position of a monosubstituted benzene ring. The 3,5
positions are equivalent, and are termed meta positions. The position to which the substituent is
attached is referred to as the ipso
position. Note that there is no fractional negative charge on the ipso or meta
positions.

q
Examining canonical
structure 1-4, we can see that
structure 1 is the lowest energy
structure. First, because the negative charge is on oxygen, whereas it resides
on carbon in the other three structures. But equally importantly, structures 2-4 imply that the aromaticity of the benzene ring has
been disrupted, whereas it is seen to be intact in the first structure.
Consequently, the real phenoxide anion looks more like structure 1 than any of the other structures (more of the negative
charge is on oxygen than on the ring positions), but there is a significant
fraction of negative charge on the ortho and para positions of the ring.
q
Since the
“best” canonical structure is 1, and there is not another structure of equal or about
equal energy, the resonance stabilization of the phenoxide anion, while
substantial, is not nearly as great as that of a carboxylate anion, which has
two equivalent resonance structures. Consequently, phenols are not as acidic as
carboxylic acids (pKa 5).
Separation of
Phenols by Extraction.
Since phenols are
substantially (ca. 106)
more acidic than water, they can be completely neutralized by the conjugate
base of water, hydroxide ion.
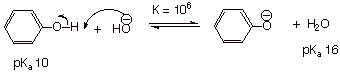
q
Consequently, they can
be separated from typical alcohols, which are not extensively neutralized by
aqueous hydroxide ion. The neutralization reaction shown above means that
phenols are converted to ionic salts (e.g., if sodium hydroxide is used as the
base, sodium phenoxide is generated), which are and insoluble in organic solvents. Typical alcohols (except for the lower molecular weight
alcohols, which are water-soluble themselves) are not neutralized and remain
soluble in the organic phase of a water/organic solvent two-phase system (e.g.,
ether/water). The separation of the two phases would then yield, after
acidification, the phenol from the aqueous phase, and the alcohol (or most
other classes of organic molecules other than carboxylic acids) from the
organic phase.
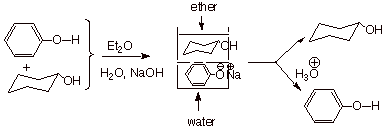
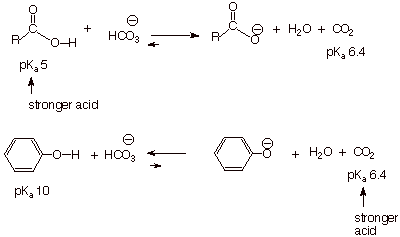
q
It is important to note
that carboxylic acids are the only
other functional group which would be completely neutralized by aqueous sodium
hydroxide and thus dissolve in the aqueous phase as a carboxylate salt. Any
other class of organic compounds, alcohols, carbonyl compounds, alkanes,
alkenes, alkynes, aromatics, organic halides, or ethers would remain in the organic
phase and be separated from the phenol. How could we separate phenols from
carboxylic acids using similar extraction procedures which, incidentally, are
far more convenient than distillation, re-crystallization, column
chromatography, etc.?
q
Obviously, we would need
a weaker base than hydroxide ion, but one which is strong enough to neutralize
the stronger class of acids, the carboxylic acids, while not extensively
neutralizing phenols. The appropriate base is bicarbonate ion, usually as
aqueous sodium bicarbonate. This converts the carboxylic acid to its anion,
which goes into the aqueous solution, allowing the carboxylic acid to be
obtained by acidification of this aqueous solution. The phenol, on the other
hand, is not neutralized, but remains in the organic layer.
An Aspirin Synthesis.
At this point in the
semester, an aspirin might be just the thing!! We can oblige, while still
keeping the subject relevant to phenol chemistry. A very common synthesis of
aspirin (acetylsalicylic acid) starts with phenol and, by means of
neutralization with sodium hydroxide, converts it to sodium phenolate. As we
have seen, this conjugate base of phenol does have substantial fractional
negative charge on its rings positions and thus it can react there, as well as at
oxygen, with appropriate electrophiles. In this case, carbon dioxide is the
appropriate electrophile.
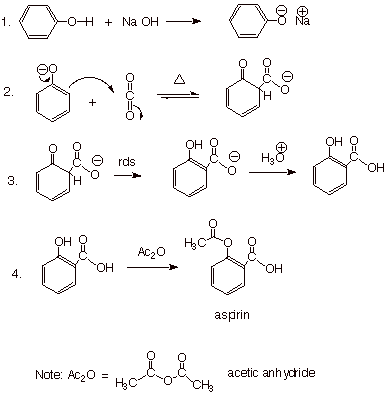
q
The second step involves
disruption of the aromatic ring, and so it is reversible and requires heating.
However, the aromaticity is restored in the third step, at which point the
reaction is no longer reversible, i.e., this isomerization is the
rate-determining step. Protonation of the product gives salicylic acid, which,
in the last step, is esterified most economically using acetic anhydride, instead of using the more expensive acetyl chloride
to make the acetate ester of the phenolic hydroxyl.
q
Although we
haven’t studied anhydrides previously, they are an acyl derivative which
is highly reactive, but not quite as reactive as acid chlorides. In this
economical synthesis of aspirin, the key organic reactants are phenol, acetic
anhydide, and carbon dioxide, all of which are relatively inexpensive chemicals
which are available in bulk.
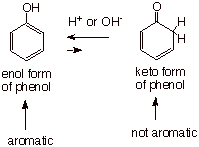
The Keto Form
of Phenol.
In a sense, phenol
is the “enol” form of a ketone (see illustration below). Although
normally the keto form of a carbonyl compound would be much more stable
thermodynamically than the enol form, in the case of phenol the
“-ene” is part of an aromatic system. This aromaticity makes the
“enol” form of phenol more stable than the keto form, in which the
aromaticity has been disrupted. Nevertheless, an equilibrium between the two
forms is presumably established in either acidic or basic media, by the same
mechanism as prevails for the normal keto/enol equilibrium. A derivative of the
keto form of phenol is actually involved in the reaction of sodium phenoxide
with carbon dioxide, in the aspirin synthesis.
Benzylic Anions, Radicals, and
Carbocations.
We have seen that
the conjugate base of phenol, the phenoxide anion, is substantially resonance
stabilized, resulting in enhanced acidity for phenols as compared to ordinary
alcohols. An entirely analogous situation applies to the corresponding anion in
which a carbon atom (with two attached hydrogens) replaces the oxygen atom of
the phenoxide anion.
q
The prototype anion of
this type is called the benzyl carbanion. A benzylic carbon, generically, is a
carbon atom which is directly attached to (but not incorporated into) an
aromatic ring. Any hydrogens attached are termed benzylic hydrogens. Toluene,
e.g., has one benzylic carbon and three benzylic hydrogens. Ethylbenzene also
has just one benzylic carbon and two benzylic hydrogens. The methyl carbon and
its three attached hydrogens are non-benzylic.
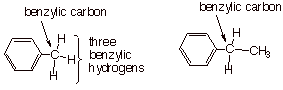
q
The resonance structures
are essentially the same as for phenoxide (see illustration below), and again
more of the negative charge is on the benzylic atom (aromaticity is disrupted
in the other three canonical structures), but there is a substantial delocalization
of negative charge to the ortho and para positions of the benzene ring.
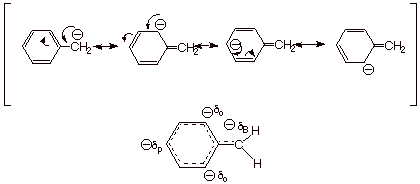
q
As a result of the
increased anion stability, the pKa of toluene (ca. 38) is much lower (it is more
acidic) than that of an alkane (pKa >50).
![]()
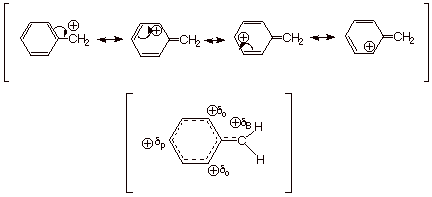
Benzylic Carbocations.
In exactly the same
way, when positive charge is generated on a benzylic carbon atom, the resulting
benzylic carbocation is highly delocalized and resonance stabilized. Again,
positive charge appears only on the benzylic (B), ortho (o), and para (p) positions
of the ring.
Benzylic Radicals.
Once again, when a
radical site is generated on a benzylic carbon atom (or indeed other benzylic atoms), the corresponding
benzylic radical is highly delocalized and strongly resonance stabilized.
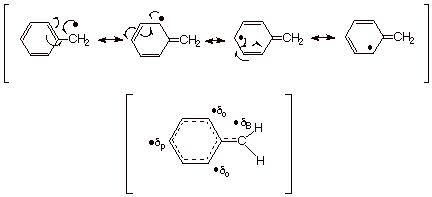
q
As a consequence of this
resonance stabilization, benzylic C-H bonds are much easier to dissociate than
typical alkane C-H bonds. They are roughly comparable to allylic C-H bonds in
their dissociation energies. For comparison, the C-H bond of ethane has a D of
98 kcal/mol.
![]()
q
Benzylic C-H bonds are
therefore easily brominated by bromine or N-bromosuccinimide under radical
conditions. Recall that tertiary and allylic C-H bonds are the only other types
of bonds that are readily substituted by the highly selective bromine atoms
involved in radical chain bromination.