Chapter 21: Reactions of Aromatics
Although benzene, as
the prototype of aromatic systems, formally has three C=C double bonds, its
reactions are quite different from those of alkenes. Whereas alkenes tend to
undergo addition reactions, especially electrophilic additions, benzene tends
to under substitution. Essentially
the preference for substitution over elimination derives from the importance of
retaining the aromaticity of the benzene ring, which would be lost if addition
occurred to one of the double bonds.
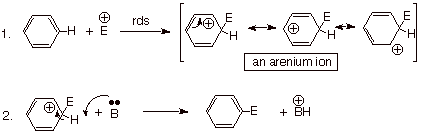
A Generalized
Mechanism for Electrophilic Aromatic Substitution.
q
Note that the first step
(sometimes called the “attack step”, is rate-determining. That is primarily because it is in this step that
the aromaticity of benzene is disrupted. Step 2, in which the stabilizing
aromaticity is restored, is naturally fast.
q
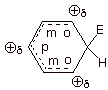
Also important is the
fact the the intermediate carbocation,
which in this context is termed an arenium ion, is itself highly delocalized and resonance
stabilized, but not aromatic. The tetrahedral carbon to which the
electrophile bonded interrupts the efficient cyclic conjugation, since it does
not have a 2pz AO to contribute to the conjugated system.
Nevertheless, there are three resonance structures which, somewhat like the
benzyl carbocation, distribute the positive charge to the ortho and para
positions of the ring.
q
We should note also the
the carbocation intermediate (the arenium ion) potentially could have reacted
with the base/nucleophile, B, at an ortho position, to give a net electrophilic
addition reaction, instead of eliminating a proton to yield the substitution
product. This kind of AdE mechanism would be exactly analogous to
the behavior ordinarily observed for alkenes. The arenium ion does not do this
because the reaction path involving proton elimination regenerates the
aromaticity of the ring, while reaction at the ortho position would have given
a product which is not at all aromatic, and would have lost even the resonance
stabilization of the arenium ion..
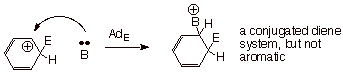
The Reactivity of
Benzene.
We have just seen
that the qualitative sense of the
reactivity of benzene (and other aromatics) is quite different from the
reactivity mode preferred in simple alkenes. But what about the quantitative reactivity, i.e., the relative rates of reaction of
benzene and, say, ethene as an example of a simple alkene?
q
As a direct result of
its aromatic resonance stabilization, benzene reacts much more slowly than alkenes with a wide variety of reagents. Recall
that stabilization of the reactant tends to increase the activation energy for
a reaction and to decrease the rate. A dramatic example is the reaction (or
rather the non-reaction) with bromine. Whereas alkenes typically react
virtually instantaneously (in real time) with bromine, benzene essentially
doesn’t react with bromine at all. [We will see shortly that bromine can
be induced to react with benzene by the use of an appropriate electrophilic
catalyst, but bromine itself is not reactive enough to undergo reaction with
benzene.]

Nitration.
The nitro group (-NO2)
is an extremely versatile substituent. Not only is this substituent important
in its own right, but it is readily converted, as we will see later, to a
variety of other substituent groups. The ability to introduce this substituent
onto the benzene, or other aromatic rings, is useful synthetically as wel as
industrially.
q
First, recall that the
nitro group is a resonance-stabilized group. There are two equivalent resonance
structures.
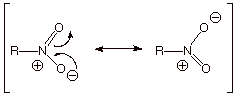
q
Also, note that in both
structures there is positive charge on nitrogen, but the negative charge is shared equally with the
two oxygen atoms. Since both structures are ionic, this group is a very highly polar group. Further, since the aromatic ring will be
attached to the positively charge nitrogen atom, the nitro group is a strongly electron-withdrawing
group (EWG). In fact, it is the
strongest of the substituents commonly encountered inorganic chemistry.
q
The nitro group can be
introduced , basically, from the nitro portion of nitric acid. However, benzene itself is too unreactive to react
directly with nitric acid, requiring a stronger acidic catalyst in order to
accelerate the reaction. Typically, sulfuric acid is used as that stronger
acid. The nitrating agent HNO3/H2SO4
is often called “mixed acid”.
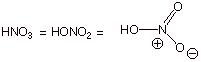
q
The detailed mechanism
of the nitration of benzene with mixed acid is given below:
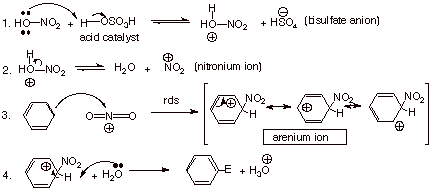
q
The usual
“two-step mechanism” for aromatic substitution is represented in
steps 3 and 4, with the “attack step” being rate determining. The
first two steps are necessary in order to generate the “active
electrophile”, the nitronium ion. As was pointed out before, nitric acid
is not reactive enough to nitrate benzene directly, but must be converted to
the stronger electrophile.
Brormination.
Bromination of
aromatic rings can also be accomplished efficiently, but, again, molecular
bromine is not a reactive enough electrophile to brominate benzene at a
convenient rate.
q
The most common catalyst
for aromatic bromination is the electrophilic catalyst ferric bromide, which is conveniently generated in situ (in place) by the reaction of bromine with catalytic
amounts of iron.
q
Although, by analogy
with nitration, one might conceive that the more reactive electrophile which
introduces bromine onto the benzene ring might be Br+, this turns
out not to be the case. The active brominating agent still contains the
catalyst; it is a “complex” of ferric bromide with bromine.
q
The detailed mechanism
for electrophilic aromatic bromination, catalyzed by ferric bromide is provided
below:
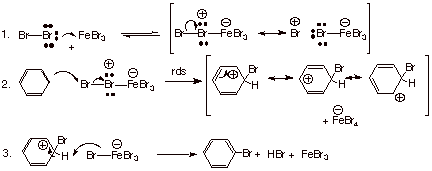
q
Recall that the
halogens, when univalently bonded, have three electron pairs. They are not very
basic or nucleophilic, but they are more nucleophilic than basic. Through one
of the electron pairs on one of the bromine atoms, a covalent bond is formed to
iron through one of the vacant orbitals in Fe+3, i.e., with ferric
bromide active as the electrophile.
This complex is a weak one and is reversibly formed, but the positive charge on
bromine makes the terminal bromine of the complex more electron deficient and
thus more reactive as an electrophile toward the benzene ring. Incidentally,
the central bromine, which also has positive charge, cannot bond to benzene
because bromine cannot readily expand its valence to four (this bromine is already
trivalent).
q
The active
electrophile, this complex between
the electrophilic catalyst and nucleophilic bromine, is more reactive than
bromine but less reactive than free Br+ would be. Apparently, rapidly than the hypothetical
dissocation of the complex to Br+.
q
Since these reactions
are not carried out in aqueous solution, the best base available to remove the
proton from the intermediate arenium ion is FeBr4-.
q
Incidentally, chlorine reacts with benzene in an entirely similar way. A few
very reactive aromatics, which have very electron-rich rings, are able to react
directly with molecular bromine or chlorine, without the need for an
electrophilic catalyst. A few very unreactive ones may require the generation
of Br++.
Friedel-Crafts
Alkylation.
A carbon atom which
is attached to a halogen atom(as in an alkyl halide) has electrophilic
character, since it represents the positive end of a substantial bond dipole.
However, alkyl halides are not very strong electrophiles and cannot, by
themselves, effect electrophilic substitution on benzene. However, in a manner
very similar to bromination and chlorination, electrophilic catalysts can be
used to enhance the fractional positive charge on that carbon atom, and thus to
enhance its electrophilicity to the point where it is indeed reactive enough to
react with benzene and other aromatics.
q
Interestingly, the
precise mechanism of Friedel-Crafts alkylation of benzene depends upon the
nature of the alkyl group in just the same way as the SN reactions
of alkyl halides with various nucleophiles do.
Mechanism of Alkylation of Benzene with Methyl Halides.
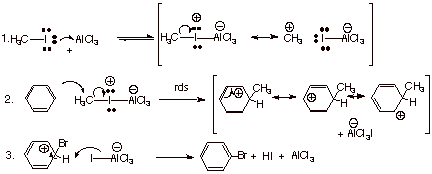
q
As in the case of
bromination/chlorination, a Lewis acid/Lewis base complex is formed between the electrophilic catalyst,
aluminum chloride (recall that Al is in Group III of the Periodic Table and
that, like boron, it has a vacant valence shell p orbital when trivalent) and
the nucleophilic iodine atom via one of the unshared pairs on iodine.
q
This complexation
generates positive charge on the iodine atom, which then also attracts
electrons from carbon, making the C-I dipole even larger. The now
more-electron-deficient methyl group then becomes a stronger electrophile than
in the uncomplexed alkyl halide.
q
Also as before, the
strongest base available to accept the proton from the arenium ion is the
negatively charged tetravalent aluminate species.
q
IMPORTANT: The above reaction represents an
electrophilic substitution (SE) on benzene, but it is an SN2
reaction on an alkyl halide. The nucleophile in this latter reaction is
benzene, which replaces the iodine substituent of methyl iodide. Step two is a
concerted loss of the leaving group AlCl3I-, along
with the formation of a bond to
nucleophlic benzene.
q
It is important to
understand that the simultaneous existence of electrophilic and nucleophilic
substitutions is not by any means an anomaly. The electrophilic and
nucleophilic roles are complementary, i.e., the role of an electrophile is to
react with a nucleophile, and conversely. So there is always a nucleophilic process that is concomitant with
any electrophilic process.
q
When benzene undergoes
substitution by, say, nitronium ion the organic chemist says this is an
electrophilic reaction because from the point of view of the organic molecule it
is, i.e., the organic molecule reacts
with an electrophile. But, from the point of view of an inorganic chemist, the
reaction could be equally well said to be a nucleophilic reaction (of benzene
as the nucleophile) with the inorganic nitronium ion.
q
The different situation
which arises in the Friedel-Crafts alkylation reaction is that both the
electrophile and the nucleophile are organic, so that even to the organic
chemist the classification as electrophilic or nucleophilic becomes ambiguous.
With respect to benzene (i.e., from the point of view of the organic molecule
benzene) it is an electrophilic substitution. But, from the point of view of
the organic molecule methyl iodide, the reaction is a nucleophilic reaction. In
such a case it is necessary to specify the reference molecule: electrophilic
with respect to benzene; nucleophilic with respect to methyl iodide.
Friedel-Crafts Alkylation of Benzene with Secondary and Tertiary Halides.
q
It should be obvious
that tertiary alkyl halides would not undergo substitution by an SN2
mechanism (steric hindrance in the pentacovalent TS). Instead, they prefer an SN1
type mechanism involving tertiary carbocations. Since benzene is a very weak
nucleophile, secondary halides also react via an SN1 mechanism.
q
The detailed mechanism
for the alkylation of benzene by tert-butyl
chloride is presented below:
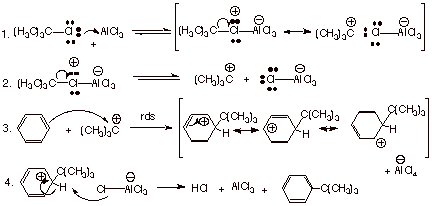
Friedel-Crafts
Alkylation By Primary Halides.
The alkylation of
benzene by primary halides proceeds in good yields, but is not normally a
useful synthetic reaction because it provides mixtures. Although the expected
mechanism for a primary halide is the SN2 mechanism, and this does
account for part of the product, a large part (usually the predominant part) of
the product results from a rearrangement of the alkyl halide/aluminum chloride
complex to give a secondary carbocation, which then proceeds to alkylate benzen
by an SN1 mechanism.
q
A typical example is the
alkylation of benzene by butyl chloride (1-chlorobutane). The SN2
mechanism provides butylbenzene (35%), but the rearranged complex affords
1-methylpropylbenzene (sec-butylbenzene).
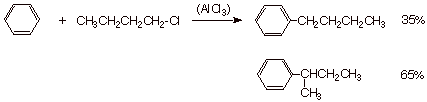
q
Although a primary
carbocation is never involved as an intermediate, the Lewis acid/Lewis base
complex has sufficient carbocation character on the primary carbon to permit a
hydride shift to give the secondary carbocation, which then alkylates benzene
via the SN1 – like mechanism.
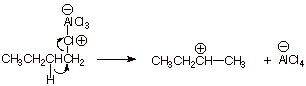
Friedel-Crafts Acylation.
Acylation of an
aromatic ring, the insertion of an acyl group onto an aromatic ring, is
accomplished using an acid halide, normally an acid chloride, in a way
analogous to that employed in Friedel-Crafts alkylation.
q
Aluminium chloride is
again required in order to facilitate the reaction, but in this case the
reaction is not catalytic. A
stoichiometric amount (equimolar amount) of this promoter is required (we will
see why).
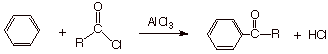
q
The product is a ketone.
q
The mechanism is always
of the SN1 type, because the acylium ion is a highly resonance stabilized cation, having both carbocation and oxonium
ion character. The carbocation center is stabilized by one of the electron
pairs of the oxygen atom.
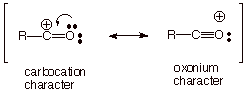
q
The mechanism of
Friedel-Crafts acylation is as follows:
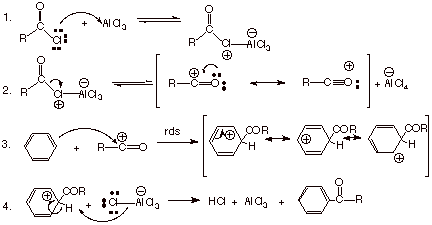
q
Although the aluminum
chloride is regenerated in step 4 of the mechanism, it immediately forms a
rather strong Lewis acid/Lewis base complex with the ketone function. The
ketone oxygen is a stronger Lewis base than either the chlorine or the oxygen
atom of the acid chloride, so that the aluminium chloride remains complexed to
the ketone and is not available for further complexing with the acid chloride.
Consequently, a mole of this promoter is needed for every mole of product
ketone formed. Incidentally, the ketone is released from the complex upon
aqueous workup, because water complexes (and reacts) with aluminum chloride more
strongly than does the ketone.
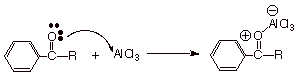
Reaction Rates and Positional Selectivity in the Electrophilic Aromatic Substitution Reactions of Monosubstituted Arenes.
We have just shown
how a variety of substituents can be introduced onto a benzene ring using electrophilic
aromatic substitution reactions. The products of these reactions are
monosubstituted benzene derivatives. Since monosubstituted arenas still have
five hydrogens attached to the benzene ring, it is obvious that they may be
able to undergo further substitution reactions, successively generating di-,
tr-, tetra-, penta-, and even hexasubstituted arenas. We want to consider the
second step in this sequence, the conversion of monosubstituted arenas into
disubstituted arenas.
q
Note that in a monosubstituted
arene, the two ortho positions are equivalent as are the two meta positions. There are therefore only three possible disubstitution products (the ipso carbon has no hydrogen attached, and normally
reaction would not occur here; in any case this would not give a disubstitution
product).
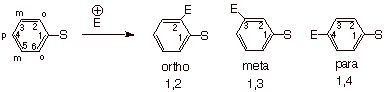
q
Besides the positional
selectivity problem (ortho vs meta
vs para), there is also the
question of how the substituent already present on the monosubstituted arene
ring affects the overall relative rate of the reaction (compared, e.g., to the
rate if that substituent were not present, i.e., compared with the rate of
reaction of benzene itself). Some substituents accelerate the rate in comparison to benzene ( in comparison to
hydrogen as the “substituent”) and others decelerate the rate. Some substituents are strongly rate
accelerating or decelerating and others are weakly so.
q
To more rigorously
discuss the question of rate effects, we need a general model of the
transition state for the rate determining step. Further, to discuss the question of positional
selectivity, we need a transition state model for all three transition states,
i.e., those leading respectively to ortho, meta, and para disubstituted
products. We would then the Method of Competing Transition States to compare
the relative rates of these three competing reactions.
q
First, we want to
develop a generalized TS model for electrophilic aromatic substitution. Recall
that the TS can be represented, using resonance theory, as a resonance hybride
of reactant-like and product-like structures at the specific geometry of the
TS. The dotted line/partial charge structure is then constructed from this and
the model characterized as carefully as possible. This includes the use of the
Hammond Principle to decide whether the character is extensive (highly
developed), moderate, or minimal.
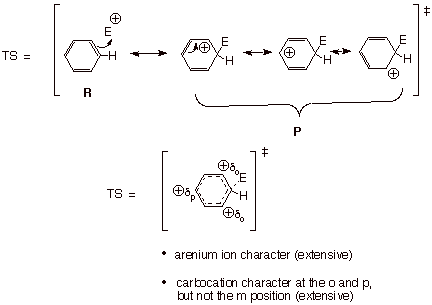
q
Since the reaction of
the electrophile with the highly stabilized aromatic ring is typically the
rate-determining step and is substantially endothermic, the TS resembles the
product of that step, which is the arenium ion. Therefore the TS has extensive
arenium ion character, which is
equivalent to extensive carbocation character at the ortho and para, but not
the meta, positions.
q
Using this predominant
character, we can now reason about the effect that a specific substituent would
have on the energy of this TS and therefore upon the rate and selectivity of
the relevant reaction of a monosubstituted arene having this substituent
present on the ring. Specifically, substituents which stabilize carbocation
character will lower the energy of the TS and accelerate the reaction rate.
These substituents are called electron-donating groups (EDG’s). Groups which destabilize a carbocation center are
electron-withdrawing groups (EWG’s), and these raise the energy of the relevant TS and retard the
reaction rate.
q
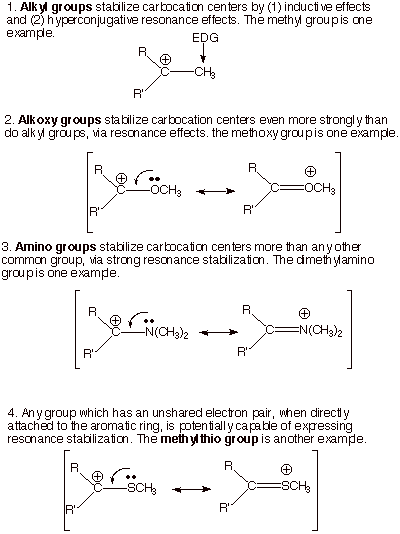
We already know some
groups which stabilize carbocation centers (EDG’s). Alkyl groups, in particular, are common but rather effective groups in stabilizing
carbocations and carbocation character in a TS. We have also seen that oxygen atoms attached to a carbocation center stabilize that
center in a highly effective way, using one of the unshared pairs on the oxygen
atom. Nitrogen atoms are
potentially even more effective than oxygen at stabilizing carbocation
character. Any group which has an unshared electron pair is capable of
stabilizing a carbocation center or carbocation character.
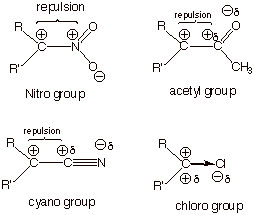
Electron-withdrawing Groups.
Electron-withdrawing
groups ( EWG’s) are groups which, when attached to a carbocation center,
destabilize the carbocation. These are typically inductive effects, because
resonance effects always produce stabilization (or no effect), but never
destabilization.
Applying the Method of
Competing Transition States to the Rationalization of Positional Selectivity in
Electrophilic Substitution of Monosubstituted Arenes.
q
Consider, first, an
electrophilic subsitution reaction (nitration) of toluene as a specific
example. Recall that the arenium ion-like TS has extensive carbocation
character at the positions ortho and para to the entering electrophile (not to the substituent, the methyl group).
q
Use the generalized
TS model previously developed, but
substitute the nitro group for the general electrophile, E+.
q
Construct three TS
models, one with the entering electrophile para to the substituent methyl
group, one with the electrophile meta to the methyl group, and one with the
nitro group electrophile ortho to the methyl substituent.
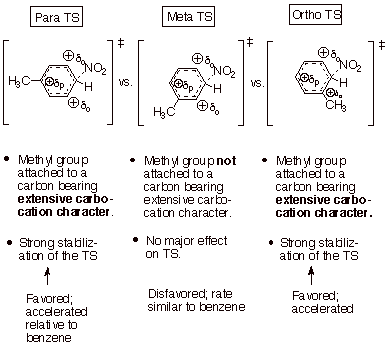
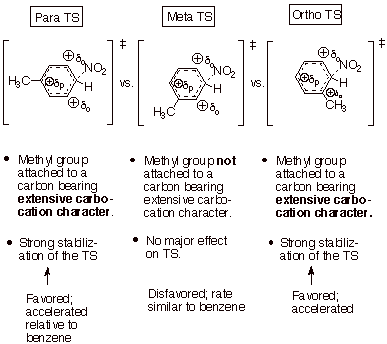
q
We see that one TS, the
meta TS does not place
carbocation character on the ring carbon atom directly bonded to the
substituent methyl group.
Consequently, the methyl group is unable to stabilize the carbocation center in
a major way. The rate of substitution of the nitro group at the meta position is rather similar to that for a position in
benzene.
q
In contrast, both the para and ortho
TS’s generate extensive carbocation character on the ring carbon directly
attached to the methyl group, which can then exert a strong stabilizing effect
on the TS. The result is that substitution at both the ortho and para positions
is accelerated relative to benzene, and also relative to the meta position. Positional selectivity therefore favors
o,p over m substitution.
q
The results for the
nitration of toluene are illustrated below:
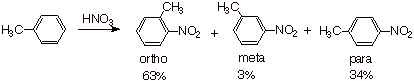
q
The main result is that ortho and para
substitution is favored over meta.
This is a general result for any EDG.
q
The greater amount of ortho than para
product can be explained statistically, since there are two equivalent ortho positions in toluene, but only one para position. On a “per position” basis, the ortho
and para sites are equally reactive. Generally, both ortho and para
products are formed in major amounts (and would need to be separated), but the
meta product is typically rather negligible.
q
In terms of reaction
rates, toluene reacts about a thousand times faster than benzene. You may have noticed that sulfuric acid (mixed acid)
was not used in the results specified above. Toluene is so much more reactive
than benzene, that the use of sulfuric acid as a more acidic catalyst is not
necessary. Nitric acid is acidic enough to act as the acid catalyst for
generating the nitronium ion.
q
To compare the relative
rate of nitration of toluene with that of benzene, we should use the Method of
Competing TS’s. Since meta
substitution is only a minor component, most of the rate acceleration comes
from reaction at the para and ortho positions. Since the rates of reaction at the latter
two positions are about equal, we need only compare one of these with benzene.
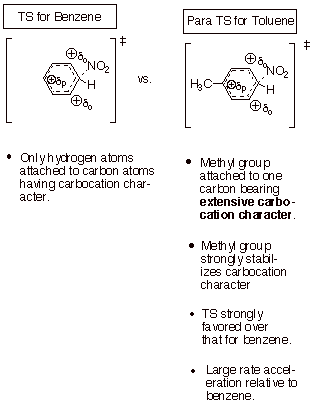
q
You could go through the
same TS arguments for the methoxy or the dimethylamino substituent. This would
be good practice. In both of these latter cases, ortho, para substitution is strongly favored and the rate
enhancements relative to benzene are even greater. For example, anisole
(methoxybenzene) reacts at a rate approximately a million times faster than
benzene. In the case of bromination of anisole, no ferric bromide catalyst
need be used. Anisole reacts rapidly with molecular bromine, to give the
mixture of o- and p-bromoanisole.
The Effect of
Electron-Withdrawing Groups (EWG’s) on Positional Selectivity and
Relative Rates
q
EWG’s exert
effects on positional selectivity and relative reaction rates exactly opposite
to those for EDG’s: meta substitution is preferred and reaction rates
are retarded, usually very strongly retarded.
q
The rationalization of
the positional selectivity and relative rate effects is parallel to that
discussed for EDG’s, except that now the EWG’s destabilize
carbocation character and especially
strongly so when the carbocation character resides on the ring carbon to which
the EWG is directly attached.
q
The Method of Competing
TS’s can again be used to rationalize positional selectivities. In this
case, for brevity, only the para
and meta TS’s will be
compared, since whatever conclusions apply to the para TS would also apply to the ortho TS.
q
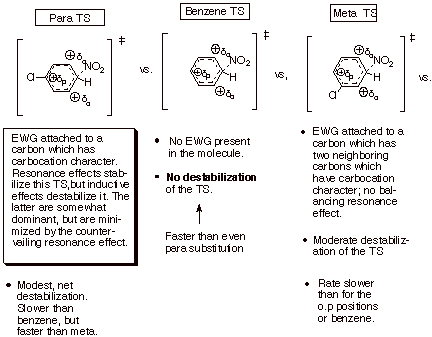
We see that the
destabilization of the TS is greater for the para (and ortho)
TS than for the meta TS, so meta substitution is favored.
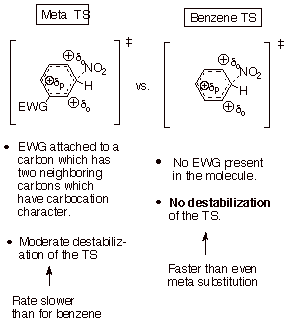
q
However, the meta TS is
also substantially destabilized relative to benzene, because the
electrostatic repulsion between the EWG’s dipole and the positive charges
on the two nearby, but not directly attached, ring positions is still
substantial. Consequently, the rate of reaction, even at the meta position, is
much slower than the rate of the corresponding reaction with benzene.
Halogen Substituents.
The halogen
substituents essentially are rule-breakers. While the great majority of all
substituents conduct themselves according to the rules, i.e., EWG’s
are meta-directing and rate-retarding, while EDG’s are o,p-directing and
rate enhancing, the halogen
substituents are o,p-directing and rate-retarding.
q
Since the halogens
decrease the rate of electrophilic aromatic substitution, they are by
definition EWG’s. In an overall
sense they decrease the reaction rate at all positions by destabilizing carbocation character.

q
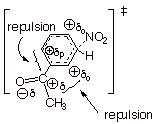
This destabilization
occurs as a result of the large C-Hal dipole moment, which has the positive end
of the dipole closer to the positive charge on the ring in the TS.
q
However, their overall
destabilization of the TS by halogens is less at the o,p positions than at the meta position, because they have a modest resonance
stabilizing effect on the carbocation character in the arenium ion-like TS when
present at the latter positions. Remember that, unlike electrostatic or
inductive effects, resonance stabilization of a carbocation center can be
effective only when the substituent is directly attached to the center which
has carbocation character. Therefore,
since positive charge occurs only at the o and p carbons of the
ring, only in this position can the halogen exert its resonance stabilizing
effect.
q
However, the resonance
stabilization is less than the inductive destabilization, so that the halogens
are overall EWG’s.
q
The major products of
the nitration of chlorobenzene are the ortho and para isomers.
Conversions of
Disubstituted Arenes to Trisubstitued Arenes.
The basic question
at issue is as follows. When two substituents are present in the reactant
arene, to what position is the entering electrophile preferentially directed?
We will consider two characteristic examples.
q
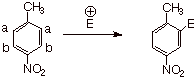
Case I.: The two substituents are para to each other, and
are of opposite directional tendencies.
In the example given, 4-nitrotoluene, the methyl group is an EDG and the nitro
group is an EWG. In this case, both groups would prefer for the electrophile to
enter at position a , which is meta to the nitro group and ortho to the methyl group. Consequently, there is no
problem in predicting the position that the new electrophile will enter. Incidentally, the electrophile could be
any of the electrophiles which we have studied in this unit (nitration,
bromination, F-C alkylation and acylation, etc.)
q
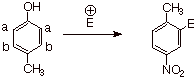
Case II. The two substituents are para to each other and
are of the same directional tendency (in this instance, both are
o,p-directors). For example, in
p-methylphenol, the hydroxyl group would tend to direct the electorphile to
position a , while the methyl
group would tend to direct it to position b, since both groups are o,p-directors. However, as we
have learned, the –OH group is a more powerful EDG than the methyl group,
so the electrophile is exclusively directed to position a.