Unit 4: Free Energy Relationships
The Hammett Equation.
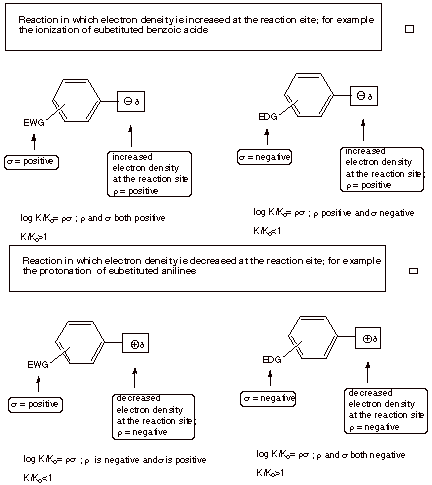
The correlation of reaction
equilibria and rates with changes in structure is a major goal of chemistry. In
organic chemistry, the change in an equilibrium constant, K, or a rate
constant, k, which results from
the substitution of a specific group for hydrogen, the so-called substituent
effect, is of special interest.
Professor L.P. Hammett, of Columbia University, systematized much of the
research in this area by defining
a quantity s (the substituent constant) for any given substituent.as follows:
![]()
where Ko is the acid dissociation
constant for the ionization of benzoic acid and K is the acid dissociation
constant for the ionization of a substituted benzoic acid with a given substituent
at a given position on the aromatic ring. Since log Ko is directly
related to the standard free energy change accompanying the ionization of
benzoic acid (via DGo
= -2.303RTlog Ko ), and log K is directly related to the standard
free energy change accompanying the ionization of the substituted benzoic acid,
the substituent constant is actually related to DDGo, the difference in the free energy changes for the two
ionization processes, i.e., a measure of the substituent effect expressed in
terms of a free energy quantity. Since the K’s depend somewhat upon the
temperature and critically upon the nature of the solvent, s is
defined specifically for water at 25 oC. Also, since the magnitude of the
substituent effect depends upon the position of the subsituent upon the
aromatic ring, there are different substituent constants for para, meta, and ortho substituents. Typically, these
are distinguished as sp
, sm , and so
. If the ratio (K/ Ko) >1, i.e.,
the substituent has increased the acidity of the benzoic acid, s is
positive. Such a substituent is considered to be an electron-withdrawing
group (EWG),
because electron density is increased at the reaction site in the product
benzoate anion, and an EWG will favor this change by withdrawing electron
density away away from the reaction site. Groups such as m-Cl, p-Cl, m-NO2,
p-NO2, etc. which have relatively large dipole moments oriented with
the positive end directed toward the reaction site are EWG’s. On the
other hand, electron donating groups (groups which tend to increase the
electron density near the reaction site) disfavor the ionization to a
negatively charged ion and have K/Ko<1. These groups have
negative sigma values. These include alkyl groups (at both the meta and para positions), para alkoxy groups, and p-amino groups. The Hammett
substituent constants for many hundreds of substituent groups have been
measured and tabulated. It should be noted that, by definition, Hammett
substituent constants are relative to hydrogen as a basis of comparison.That is
to say, sH = 0.0.
HAMMET SUBSTITUENT CONSTANTS
Hammett
sconstants are based upon the acid dissociation of
benzoic acid and m- and p-substituted benzoic acids in water at 25 oC.
The Ka’s of some of these acids are given in the table below:
|
Substituent (X) |
Ka |
pKa |
sp |
EWG or EDG |
|
p-OCH3 |
3.5x10-5 |
4.46 |
-0.27 |
EDG |
|
p-CH3 |
4.3x10-5 |
4.34 |
-0.17 |
EDG |
|
H |
6.46x10-5 |
4.19 |
0.00 |
Standard |
|
p-Cl |
1.10x10-4 |
3.96 |
0.23 |
EWG |
|
p-NO2 |
3.90x10-4 |
3.41 |
0.78 |
EWG |
The
sigma values are derived from the equation below:
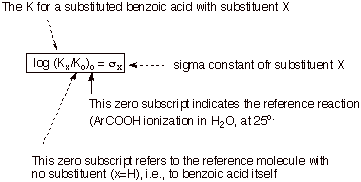
Note that is X=H, the sigma value is exactly zero, by definition. Also note that EDG’s have negative values of sigma because these substituents cause a decrease in the ionization constant (we will see why). EWG’s have positive values of sigma, because they cause an increase in the ionization constant relative to that of parent benzoic acid. These constants not only give us an idea of whether a given substituent is electron-withdrawing or electron-donating relative to hydrogen, but they also give us a qualitative and quantitative sequence of EWG-power or EDG-power. Please not, as we will see, that a para X has a different sigma value that a meta X. That is why in our first table we specified the para positioning of the substituents. Ortho substituents are not usually treated within the context of the Hammett equation because steric effects can interfere with the purely electronic effects, and these steric effects are highly variable from reaction to reaction.
The
Hammett Equation (given below) then asserts that these same substituents will
have effects upon the equilibrium or rate constants for any other reaction
which linearly parallel those in the benzoic acid ionization. The
proportionality constant (i.e., the slope of the line) r, will vary with the particular reaction under study, but its magnitude
reveals the degree of sensitivity the reaction has to substituent effects. If
it is greater than 1, it is more sensitive to substituent effects than the
standard benzoic acid ionization. Here is is appropriate to note that the rho
value for benzoic acid ionization is, by definition, exactly 1.000. If the rho
value for a given reaction or equilibrium is less than 1, it is less sensitive.
On the other hand, if it is negative, it indicates that substituents affect the
reaction in the opposite way from the standard reaction, i.e., EWG’s slow
the reaction rate or suppress the equilibrium and EDG’s accelerate the
rate or favor the equilibrium. Since the benzoic acid ionization generates a
negatively charged product, and this corresponds to a positive rho value (by
definition), a negative rho corresponds to the development of position charge
(or the quenching of negative charge).
Para vs Meta
Subsituents: Resonance and Inductive Effects. Substituents exert the electronic
effects expressed in Hammett correlations via two main types of effect,
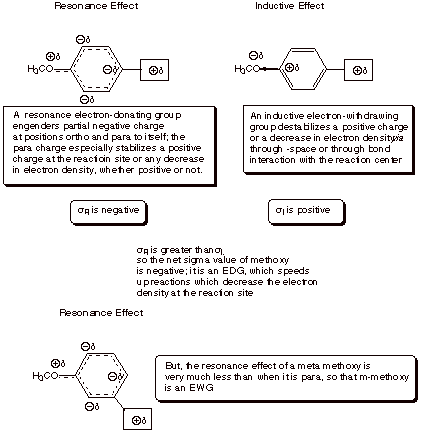
resonance and inductive effects. To illustrate the resonance effect, consider
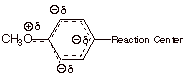
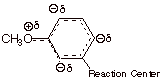
the p-methoxy substituent. Because
of a resonance interaction between a filled 2p AO on the directly attached oxygen
and the pi system of the ring (via
the ipso carbon), the methoxy substituent places a substantial amount of excess
electron density (negative charge) upon the ring. However, this charge is
selectively placed upon the ortho
and para positions of the ring, as
opposed to the meta position.
Consequently, of the methoxy group is para to the reaction center, it generates negative charge upon the ring
carbon directly attached to the reaction center’ attachment. This is the
main effect of the p-methoxy
group, which makes it a rather strongly EDG. By the way, a small effect of this
resonance is felt even when the methoxy is meta to the reaction center, because of the negative
charge generated at positions ortho
to the reaction center. Since these centers of negative charge are further away
from the reaction site, the effect is much smaller, and the m-methoxy group is
actually EWG. This is the result of its electron withdrawing inductive effect.
The para substituent constant is
considered to be a sum of the resonance sigma value and the inductive sigma
value, while the meta sigma value
is the sum of the inductive sigma and a fraction of the resonance sigma.
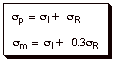
As
an example, the meta sigma value
of chlorine is 0.37, while the para
sigma is only 0.22, i.e., both substituents are EWG, but the meta one is more strongly so. Note that the inductive
effect is dominant for the halogens., in part because they are poorer electron
donors via the resonance effect than is oxygen. Also, the meta sigma of methoxy is +0.11 (it is EWG), while the para sigma is –0.27 (it is EDG). Strong evidence
exists that the inductive effects in the meta and para positions are very similar. So for para methoxy, the resonance effect is dominant over the
inductive effect, but both are significant. For an amino group, the electron
donating power is even greater. Note also that the sigma for p-SCH3 (0.00) is less negative than
that for p-methoxy, even though
the inductive effect of sulfur is surely less than that of oxygen. This is
apparently because of an orbital mismatch; that is, the larger 3p AO of sulfur
overlaps less efficiently with the smaller 2p AO of the attached ring carbon,
resulting in a much smaller resonance effect in the case of sulfur
substituents. For the same reason, a p-fluoro subsituent (+0.06) is less strongly electron
withdrawing than a p-Cl )+0.23) or
Br substituent.
Hammett
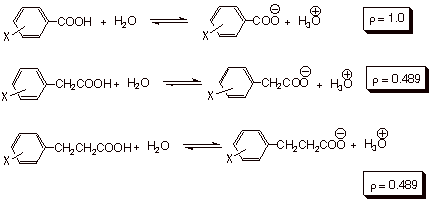
further observed that if log (K/ Ko) for the ionization of other
acids was plotted versus these sigma values, reasonably linear plots were obtained.
For example, if the log (K/ Ko) data for the ionization of meta and para substituted phenylacetic acids
in water at 25 oC was
plotted versus his sigma values a linear plot of slope 0.489 was obtained.
Similarly, if the data for ionization of substituted phenylpropionic acids was
plotted vs sigma, a linear plot of
slope 0.212 was obtained.
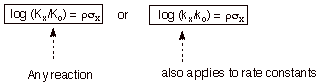
Hammett therefore proposed the equation now known by his
name:
![]()
where
log (K/ Ko) represent the log of the relative equilibrium constants
for any reaction, with the K for the substituted reactant in the numerator and
the Ko for the unsubstituted version in the denominator. The value
of the proportionality constant, rho, depends upon the specific reaction being
studied, and was termed the reaction constant. Essentially, the Hammett
equation proposes that the relative free energy changes occasioned by the
changes in substituent are proportional the same changes in the benzoic acid
series, or for that matter to any other series of equilibria. The magnitude of
rho, in effect, determines how large the free energy changes are in a reaction
series, that is, how large the substituent effects are. If K>1, the
substituent effects are larger than in the standard benzoic acid ionization. If
K<1, the substituent effects are smaller than in the standard reaction. We
can easily see that the substituent effects are smaller for the arylpropionic
acid series than for the arylacetic acid series, than for the standard
reaction. Essentially this is because the change in electron density occasioned
by the loss of a proton for the carboxyl group, although the same in each of
these reaction series, is closest to the substituent and the aryl ring in the
standard series and most remote in the arylpropionic acid series. The
substituent effect is thus least in the latter series because of the remoteness
of the reaction site from the substituent and the aryl ring.
In
the ionization of phenols, the negative charge is generated at a benzylic-type
position, i.e., it is generated on an atom (oxygen) which is directly attached
to the ring. The rho value for the ionization of substituted phenols in water
at 25 oC is, correspondingly greater than that for the standard
benzoic acid ionization (2.1)
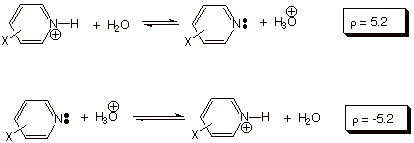
When the reaction site is actually in the aromatic ring,
the substituent effects, as reflected by the rho value, are even larger than in
the standard reaction series. This is seen in the ionization of substituted
pyridinium ions in water at 25 oC, where r = +5.2. Incidentally, the second equation below
indicates that the rho value for the reverse of a reaction has the opposite
sign, but the same magnitude as for the forward reaction. In general reactions,
like the standard reaction, in which electron density is increase at the
reaction site, have positive rho values (posite rho may be taken as a positive
increase in electron density). Reactions in which the electron density at the
reaction center is decreased in going from reactant to product have negative
rho values. The seoncd equation
below is an example of the latter kind, in which a positive charge is generated
at the reaction site. From the first equation below we should note that
electron density increases do not require the development of a negative charge.
The removal of a positive charge is equally as good at increasing electron
density.
We have seen that rho values depend quantitatively upon the distance of the electron density change from the aryl ring. They also depend, of course, upon the magnitude of the electron density change. The development of partial negative charges is best seen in a context of the correlation of reaction rates with substituent constants (see below). However, a similar effect can be seen in the dependence of the magnitude of rho, for a given reaction, upon the nature of the solvent. As we have seen, the rho value (by definition) for the standard reaction series (in water) is 1.0. If the solvent is changed to methanol, the rho value increases by about 50%, to 1.54. In solvent ethanol it increases to almost twice the value in water (1.96). In acetonitrile, a further increase to 2.8 is observed. Clearly, solvation has a major effect on the magnitude of substituent effects, being largest when the carboxylate ion is not as highly solvated by hydrogen bonding. In effect, hydrogen bonding to the carboxylate ion partially quenches the unit negative charge on the carboxylate anion. Consequently, the rho value is least in water, the best hydrogen bonding solvent of all.
Correlation
of Reaction Rates with Hammett s Values. In an entirely similar way, relative rate constants for various
reaction series can be correlated with substituent constants. In this case, of
course, the free energy changes referred to are between reactant and transition
state. Once again, if electron density is increased in the TS relative to the
reactant, positive rho values are expected, i.e., electron withdrawing groups
will accelerate the reaction. If electron density is decreased, EDG’s
will speed the reaction. The Hammet equation as applied to relative reaction
rates would be expressed as follows:
![]()
where,
as before, k is the rate constant for the substituted version of the reactant
and ko is the rate constant for the unsubstituted version of the
reactant (X = H). As before, larger rho values can correspond to electron
density changes which are closer to the aryl ring, but in the case of
transition states, also to larger changes in electron density. This is because
transition states can have partial charges.
The
Substituent Constants. It is generally understood that substitutents exert their effects by
means of two distinct electronic mechanisms, resonance effects and inductive
effects. The latter are essentially electrostatic attractions or repulsions
resulting from the dipole moment of the ring C-X bond and its differential
interactions, both through space and through bonds, with the reaction centers
in the reactant and in the product or transition state. Resonance effects are
exerted through the increase or decrease in pi electron density in the aromatic
ring as a result of pi overlap between the 2p AO on the directly attached ring
carbon and an orbital (usually a p AO) on the substituent. Both inductive and
resonance effects are somewhat position specific, but inductive effects tend to
be quite similar for the same group when situated in either the meta or para
position. In sharp contrast, resonance effects are very small for meta
substituents, since partial charges induced by resonance are specific to the
positions ortho and para to the substituent. Therefore resonance effects are
especially large for para substituents. The following equations express these
relationships:
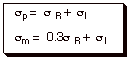
As a rough approximation, the resonance sigma
parameters can be obtained by just subtracting the meta sigma values from the
para sigma values.
Hammett Substituent Constants
|
Substituent |
sp |
sm |
sp - sm (ca. sR) |
|
NMe2 |
-0.83 |
-0.21 |
-0.62 |
|
OMe |
-0.27 |
+0.12 |
-0.39 |
|
CH3 |
-0.17 |
-0.07 |
-0.10 |
|
H |
0.00 |
0.00 |
0.00 |
|
F |
+0.06 |
+0.34 |
-0.28 |
|
Cl |
+0.23 |
+0.37 |
-0.14 |
|
CO2Et |
+0.45 |
+0.37 |
+0.08 |
|
CH3C=O |
+0.50 |
+0.38 |
+0.12 |
|
NO2 |
+0.78 |
+0.71 |
+0.07 |
|
NMe3+ |
+0.82 |
+0.88 |
+0.06 |
where
the resonance contribution to the overall sigma constant is represented as sR and the inductive contribution as sI. Groups which are electron donating
in a resonance sense are of two sub-types: (1) those, such as –Cl, -OR,
SR, and NR2 which have an unshared electron pair on the atom
attached to the ring, which can donate electrons to the ring via resonance (2)
alkyl groups, which can donate electrons via hyperconjugative resonance. These
groups, being electron donors, therefore have negative values of sR. Those substituents in the first
group have positive values of sI,
since their sigma bond dipoles are oriented with the positive end nearest the
ring. There is therefore a tendency of these two effects to operate in opposite
directions. For nitrogen and oxygen groups, where resonance effects are the
strongest, the resonance effect is dominant over the inductive effect, and the
net sigma value negative (EDG’s). The electrons on nitrogen in the amino
functions, such as amino or dimethylamino, are less tightly held than those on
oxygen in the oxygenated substituents such as hydroxy or methoxy, so the
resonance effect is larger for oxygen-center substituents. Also, the sigma bond
dipole, and therefore the inductive effect, is diminished in the case of nitrogen
as compared to oxygen. Since resonance effects are larger and inductive effects
are smaller for nitrogen, the amino substituents are more strongly
electron-donating than the alkoxy substituents. It may be interesting to think
about why the sulfur substituents are less strongly donating that the
corresponding oxygen substituents (there is an orbital size mismatch between
the 3p orbital on S and the 2p orbital on carbon, leading to less efficient
overlap).
Hammett-Brown
s+ Constants. In the case of reactions in
which positive charge is generated on a benzylic carbon (as in an SN1
reaction of certain benzylic halides or tosylates) or directly upon the
aromatic ring (as in electrophilic aromatic substitution), the resonance
effects of substitutents are rather strongly enhanced. In such cases, such that
standard Hammett sigma parameters are longer valid. In particular, the rates of
reaction of substrates having para electron donor groups such as methoxy, methylthio,
dimethylamino, and even alkyl groups are much faster than would be predicted by
the Hammett sigma values. Even para halogen substituents , which are resonance electron
donor substituents, are much less rate retarding than would be expected from
Hammett sigma values. Amazingly, para fluoro substitutents can even be modestly rate
enhancing. These enhanced resonance effects apparently arise as a consequence
of the direct resonance interaction of the positively charged reaction site
with the para
group via
quionoidal resonance forms such as those shown below. Since these interactions
are impossible when the substituent is
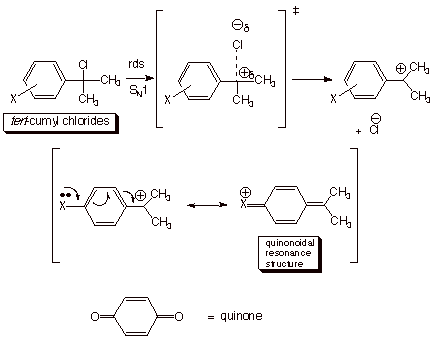
attached
to the meta position, and are not operative for even a para resonance-electron withdrawing
substituent like nitro or acetyl, the sigma values of these latter kinds of substituents are still
valid. By plotting the data for the solvolysis of tert-cumyl chlorides and
establishing the regression line on the basis of the meta substituents and the
para resonance-EWG substituents, H.C. Brown derived a new set of sigma
constants for the para resonance-EDG substituents, and designated them as s+ values.
|
Para Substituent |
s Value |
s+ Value |
|
-NMe2 |
-0.83 |
-1.7 |
|
-OCH3 |
-0.27 |
-0.78 |
|
-SMe |
0.00 |
-0.60 |
|
-CH3 |
-0.17 |
-0.31 |
|
-Cl |
+0.23 |
+0.11 |
|
-F |
+0.34 |
-0.07 |
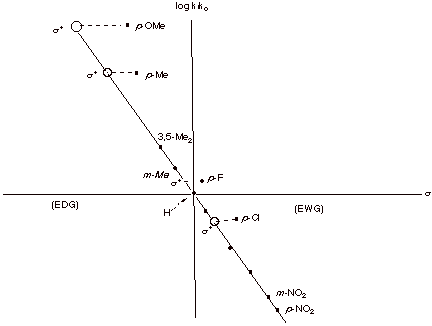
These same values are useful for the correlation of data
for electrophilic aromatic substitution, where analogous quinonoidal resonance
interactions are feasible.
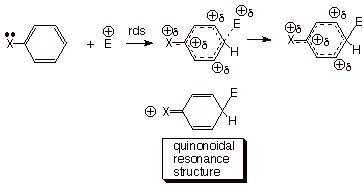
Rho Values for Various Electrophilic Substitutions. The rate-determining step for most electrophilic substitutions is the attack step in which the electrophile reacts with the aromatic to form the arenium ion. The degree to which the TS for this reaction resembles the arenium ion is a major determinant of the size of the rho value for a particular type of electrophilic substitution. For bromination, the rho value is quite large (-12.1), indicating a TS which strongly resembles the arenium ion. This is consistent with the accepted mechanism, in which neutral bromine is the (relatively mild) electrophile, so that the attack step is strongly endothermic. In Friedel-Crafts acylation, where the active electrophile is the acylium cation, the rho value decreases to-9.1. The rho value for nitration in acetonitrile is –6.2.
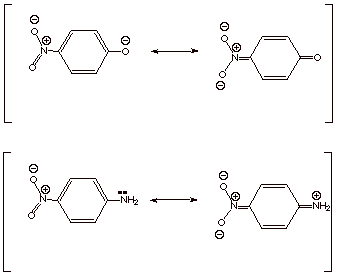
s - Values. Analogous enhanced resonance
effects are encountered for para resonance-electron-withdrawing substituents when
negative charge is generated on a benzylic atom (as in the ionization of
phenols to phenoxide ions) or upon
the aromatic ring (as in nucleophilic aromatic substitution). These same
substituent constants, called s -,
are also
applicable to the ionization of anilinium ions to substituted anilines, where a
negative charge is not developed, but the strongly resonance electron donating
amino group of the product interacts via the typical quinonoidal resonance
interaction with resonance EWG’s present at the para position.
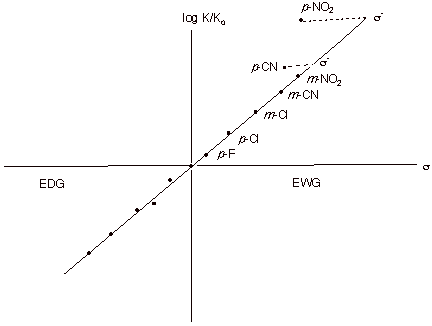
A typical Hammet plot for the dissociation of phenols is shown below. The regression line is defined by all of the resonance-electron-donating groups and by meta groups whether they are EDG or EWG. Only the para resonance-EWG groups such as nitro, cyano, and carbethoxy deviate, and their deviations are quite large. Whereas the sigma value for p-nitro is 0.78, the s- value is 1.27! On the other hand, the sigma value for meta nitro does not require any change, i.e., the s- value is the same as the s value.
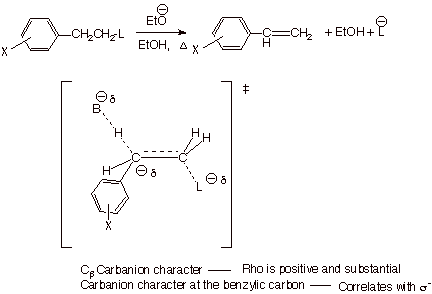
Detecting
the Cb Carbanion Character in E2
Eliminations. We
have previously developed a TS model for E2 eliminations in which the TS has
both alkene character and Cb carbanion character.
Through the study of the rates of E2 elimination of a series of m- and p-substituted
b-phenylethyl chlorides and the correlation of the
rates with the Hammett-Brown s+ constants, the
projection that carbanion character is developed on the beta carbon can be
experimentally affirmed. The observed rho value is +2.61. When the leaving
group ability is diminished by using fluorine as the leaving group, the rho
value increases to +3.12, indicating that carbanion character is now even
greater at the beta carbon. Finally, when the leaving group is
trimethylammonium ion (remember the Hoffmann elimination?), the carbanion
character increases further to +3.8. This can explain why Hoffmann-type
eliminations do not give the more stable alkene as the predominant product, but
correspond to generating the TS which has the more stable carbanion character
(primary>secondary>tertiary).