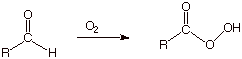Unit 5: Radicals and Radical Reactions
Introduction
Free radicals can be defined as
chemical species which have a single unpaired electron. In the important case
(for organic chemistry) of the methyl radical, the radical center is trivalent
and trigonally hybridized (Scheme 1). The sp2 hybridized carbon atom
and the three hydrogens are coplanar and the unpaired (odd) electron occupies a
2p carbon atomic orbital (AO), here arbitrarily designated as 2pz.
This singly occupied orbital is of special importance to free radical chemistry
and is often abbreviated as the SOMO
(singly occupied molecular orbital).
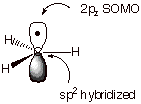
Scheme 1. Structure of the Methyl Radical
The
odd electron can have either an a or b spin, so there are two spin states which are energetically distinguishable
in the presence of a magnetic field, but which, in the absence of such an
external field, are isoenergetic. Free radicals are termed spin doublets because of the existence of these two discrete
states. In the same way, chemical species with two unpaired electrons are
called triplets, since there are
three distinguishable spin states of such a system. Species with all electrons
paired have only one spin state and are termed singlets.
Formation of Free Radicals
In
the (presumably) familiar case of radical chain reactions (such as halogenation
of alkanes), radicals are typically generated by either thermal or
photochemical homolytic bond cleavage. In the case of the chlorination of
methane, the reaction can be carried out thermally at rather high temperatures
(250 – 300 oC) by the homolytic cleavage of the Cl-Cl covalent
bond (Scheme 2). This bond has a
bond dissociation energy of 58 kcal/mol, so that a rather high temperature is
required in order to dissociate chlorine molecules fast enough to effectively
initiate the chain mechanism.
![]()
Scheme 3. Generation of Free Radicals by Homolysis of Bonds
More
commonly, in the laboratory, radical chemistry is initiated at temperatures
below 100 oC by adding a few percent of a substance (called an initiator) which has a relatively weak bond. Peroxides, which have a
weak O-O bond, are perhaps the most common choice. In the case of bromination,
since molecular bromine absorbs visible light, homolytic dissociation to
bromine atoms can be accomplished at room temperature or below by photochemical
means, i.e., by irradiation.
Radical Stabilization
An examination of the bond dissociation energies, D,
of the C-H bonds of methane, ethane, and other alkanes, it becomes evident that
radical centers are progressively stabilized by the replacement of one, two, or
three of the hydrogens of the methyl radical by alkyl groups. For example the
bond dissociation energies of methane and ethane are 105 and 98 kcal/mol,
respectively. The trend of diminishing D’s is continued as one proceeds
from the primary C-H bond of ethane to secondary C-H bonds (such as occur in
propane, butane and other alkanes: 95 kcal/mol), and on to tertiary C-H bonds
(92 kcal/mol). Much of this stabilization is considered to result from resonance
stabilization of the radical, as a result of delocalization of the odd electron
so that it is no longer required to be fully localized on the radical carbon
center (as in the methyl radical). Spectroscopic and theoretical results
indicate, e.g., that ca. 15% of
the odd electron in the ethyl radical is delocalized onto the (three) beta
hydrogens. This kind of resonance stabilization (shown in Scheme 3) is often
termed hyperconjugation, since the
resonance stabilization effects which it engenders are weaker than in the case
of conjugation (which involves only pi type orbitals, not sigma orbitals as in the case of the
beta C-H bonds).
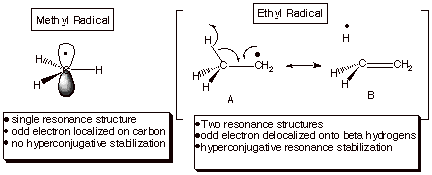
Scheme 3. Hyperconjugative Resonance Stabilization of Primary, Secondary, and Tertiary Alkyl Radicals
Conjugative resonance stabilization, which involves only the relatively weaker pi bonds, is another means of stabilizing radicals which is even more effective than hyperconjugation. The allyl and benzyl radicals are protoype examples of conjugative resonance stabilization (Scheme 4). The homollytic dissocation energy of an allylic C-H bond of propene, e.g., which generates the allyl radical is only 87 kcal/mol, making these bonds more easily dissociated than even tertiary C-H bonds. The D’s of benzylic C-H bonds are very similar to those of allylic C-H bonds.
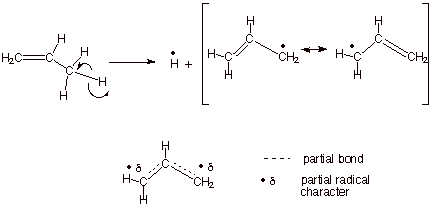
Scheme 4. Allylic Resonance Stabilizations
A
novel means of stabilizing radicals is via three electron bonding. Although it is well known that optimally
strong bonding invoves two electron bonds, in which the bonding molecular
orbital (BMO) is fully occupied and no antibonding MO’s (ABMO’s)
are occupied, it has been noted in a previous unit (Unit 1), that net
stabilization nevertheless results when the BMO is doubly occupied and an ABMO
is singly occupied. This is a fairly common situation with free radicals,
especially when the radical center (be it carbon or any other atom) is attached
to another atom which has an unshared electron pair. This latter atom can
supply two electrons toward pi
bonding, while the radical center supplies the third electron.
Persistent and Stable Free Radicals
The ultimate challenge in attaining an isolably stable
free radical, or even one that persists in solution but is difficult or
impossible to isolate, is the coupling of two radicals to afford a dimer
(Scheme 5). This typical mode of radical reaction results in the formation of a
covalent bond without the necessity of breaking any bonds. It is consequently
thermodynamically very favorable, at least for simple radicals, and also
extremely fast. The equilibrium between the monomeric radicals and the
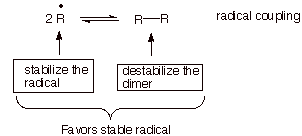
Scheme 5. Showing Approaches to Obtain a Stable Radical
corresponding dimer can
obviously be influenced in favor of the radicals by providing for one or more
kinds of radical stabilization. However, most stable and even persistent
radicals require more than just radical stabilization by electronic means. The
additional impetus required for full stabilization is typically provided by
destabilization of the dimer, in particular by steric means. A case in point is the persistent
triphenylmethyl radical, which was the very first radical to be observed
(Gomberg, 1900). This radical can be easily generated by the treatment of
triphenylmethyl chloride (“trityl” chloride) with a metal such as
silver (Scheme 6). The presence of the trityl radical in solution is easily
detected by specitroscopic means (electron spin resonance) and by its yellow
color. It is also quickly oxidized in air to the corresponding peroxide. The
persistent (long-lived but not readily isolated) trityl radical is in
equilibrium with its dimer (an interesting structure, see below). Although the
dimer is the predominant form at room temperature (ca. 98%), the radical is
present to the extent of 2% in the equilibrium. The persistent stability of
this radical is the result of a combination of effects, including conjugative
stabilization of the radical by benzylic resonance (delocalization upon three
phenyl rings) and the profound steric destabilization which would be present in
the normally expected dimer, hexaphenylethane. In fact, the actual dimer
present turns out to be one in which the aromaticity of one of the benzene
rings is disrupted, so that the dimer is destabilized not so much by steric
effects, but by the loss of aromaticity. This dimer is formed by the coupling
of one trityl radical form its benzylic carbon to the para position of the other trityl radical. As might be
expected, the tris(4-methylphenyl)methyl radical ( which has a methyl
substituent at the para position
of each phenyl ring) is a persistent radical in which dimer formation is almost
undetectable.
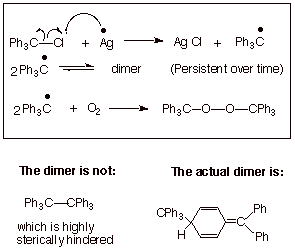
Scheme 6. The Triphenylmethyl Radical: A Persistent Radical
Still
another factor can be exploited, and is usually needed, for stabilization of a
radical to the point where it is actually isolably stable. Not only can a dimer
be destabilized by steric interactions, it can be destabilized in an even more
fundamental way, i.e., by the circumstance that the new covalent bond formed in
the dimer is of a type that is inherently relatively unstable. A very nice
example of this is seen in the 2,2,6,6-tetramethylpiperidinoxyl (TEMPO) radical
(Scheme 7). This radical exhibits an interest blend of effects which either
stabilize the radical or destabilize the dimer. In the present connection, we
not that dimer formation would require the formation of an O-O bond. In
addition, the dimer would have steric destabilization from the interaction of
the four methyl groups on each monomer unit. Finally, the radical is substantially
stabilized by three electron bonding.
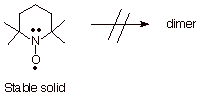
Scheme 7. TEMPO: A
Stable Free Radical
There
is also an interesting instance in which a persistent radical is formed without
the benefit of either steric destabilization of the dimer or the formation of
an inherently less stable covalent bond. The phenalenyl radical is a very
highly resonance stabilized radical which proves to be persistent in solution
(Scheme 8).
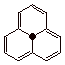
Scheme 8. The Phenylenyl Radical: A Novel, Persistent Radical
(Aromatic Radical?)
Uses for Stable Radicals: Radical Scavengers and Radical Probes
Bond homolysis forms not one but a pair of radicals.
When this radical pair is formed by solution homolysis, it is often considered
to be a caged radical pair. That is, the initially formed radical pair is
surrounded by solvent molecules, bu no solvent molecules intervene between the
pair. In order to escape this so-called cage, one or both of these radicals
must diffuse through the solvent. Of course, diffusion through most non-viscous
solvents is normally quite fast, with rate constants of about 1010 s-1.
However, the recombination of these radicals by radical coupling is also quite
rapid for most relatively simple radicals. As will be seen later, there are
sometimes means by which to track this re-formation of the reactant. In any
case, there is the potential for a competition between escape from the solvent
cage and recombination (often termed “geminate recombination”).
An interesting case in which
escape from the solvent cage and recombination can be easily tracked through
the use of stable radicals as radical scavengers is the thermal cleavage of
azobis(isobutyronitrile (AIBN; Scheme 9). It turns out that, owing the the
great thermodynamic stability of the dinitrogen molecule (it has an N,N triple
bond), both C-N bonds cleave simultaneously, in a highly concerted process, to
give dinitrogen and a caged pair of 2-cyano-2-propyl radicals. These radicals
can recombine inside the cage to form tetramethylsuccinonitrile or they may
diffuse apart to become “free” radicals. However, these free
radicals eventually encounter each other and undergo radical coupling to form
the same dinitrile. The
fundamentally interesting question is “What is the relative amount (rate)
of cage recombination and escape from the solvent cage?” This is easily
estimate by the inclusion of a stable radical such as TEMPO or DPPH (diphenyl
picryl hydrazyl; see below) or galvinoxyl, which would rapidly react with any
radicals which escape the cage, but cannot trap the caged radicals.
First,
it is essential to investigate the kinetics of the thermal decomposition of
AIBN in the absence and in the presence of stoichiometric amounts of the chosen
radical scavenger, so as to ascertain that the scavenger is not affecting the
rate of the homolysis reaction. Assuming that the scavenger does not exert any
effect upon the rate of cleavage of AIBN (and it does not), we can determine
the yield of the product dinitrile in the absence of, and in the presence of,
the scavenger. A lowering of the yield would indicate that this amount of
radical had escaped thecage and was trapped by the scavenger, instead of going
on to give the dinitrile. In a typical result, the yield of the dinitrile is
lowered to about 34% from essentially quantitative in the absence of the
scavenger. Similar results obtained for other scavengers provide credibility
for the interpretation that about 66% of the radicals escape the cage, and 34%
couple without escaping the cage. Incidentally, one consequence of this cage
recombination is that only about one-third of the AIBN is available for
initiating radical reactions. In contrast, the decompositioin of benzoyl
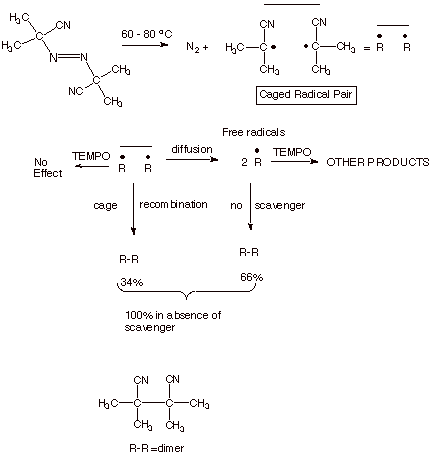
Scheme 9. The use of Radical Scavengers to demonstrate and
measure cage recombination (geminate recombination).
peroxide (often used as an
initiator for radical reactions) provides 100% of cage escape (no cage
recombination). See if you can
understand why, in the decomposition of dibenzoyl peroxide, 2.0 moles of free
radicals are produced for every 1.0 moles of peroxide which decomposes.
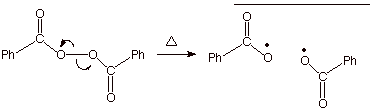
Scheme 10. Thermal decomposition of dibenzoyl peroxide is 100%
efficient in generating free
radicals.
Radical scavengers can also be used in a somewhat more elegant way, as a mechanistic diagnostic tool. A prime example is the use of TEMPO to demonstrate a radical mechanism for the formation of Grignard reagents. The general mechanism shown in Scheme 11 has been proposed for the formation of a typical Grignard reagent. To the extent that the organic radical becomes free, it should be trappable by an appropriate scavenger. As also shown in Scheme 11, 95% of the cycloheptyl radicals generated in the treatment of cycloheptyl bromide with magnesium metal in ether solvent are trapped by TEMPO as the radical coupling product. Incidentally, in the absence of a radical scavenger, the escape of free radicals usually produces significant amounts of dimer in competition with Grignard formation. It should be noted that TEMPO was included in the reaction mixture from the beginning of the reaction, and was used in stoichiometric amounts. Since Grignard formation is not a radical chain reaction, a few percent of a radical inhibitor could not be employed to suppress the reaction as a potential mechanistic test. Grignard formation is a non-chain radical reaction.
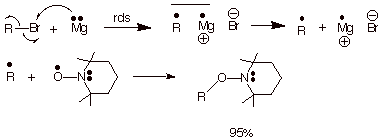
Scheme 11. Mechanism of Grignard Reagent Formation. Shown with trapping results using TEMPO in the Reaction of Cycloheptyl Bromide with Magnesium in Ether
Radical Probes
Carbon-centered radicals (as well as many other types of radicals) show a propensity for addition to carbon-carbon pi bonds. If a radical mechanism is operative in a gi ven reaction, and if an alkene pi bond is present in the molecule, an intramolecular radical addition reaction may be observed. This kind of chemistry has often been used to probe for radical mechanisms. As a rather prominent example, consider the reduction of alkyl bromides using tributyltin hydride, the proposed mechanism for which is shown in Scheme 12. Since the proposed mechanism is now a radical chain mechanism, the inclusion of a few % of a radical inhibitor to completely suppress the reaction could be used to support this mechanistic formulation. However, a more elegant approach, which
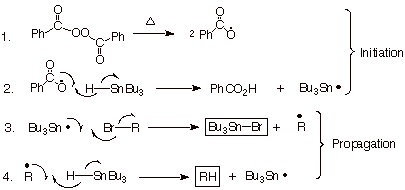
Scheme 12. Proposed Radical Chain Mechanism for the Reduction of Alkyl Halides by Tributyltin Hydride.
also establishes the specific location at which the radical site is generated, is to employ a radical probe. As an example, consider the reduction of 6-bromo-1-hexene by tributyltin hydride. If a radical site is generated at the carbon formerly bonded to bromine, the hydrogen abstraction step (step 4 in Scheme 12) which would generate 1-hexene as the product should have to compete with the intramolecular addition of the radical site to the double bond. Since the 5-hexene-1yl radical is known to cyclize to the cyclopentylmethyl radical, this should end up producing methylcyclopentane. In fact, both 1-hexene and methylcyclopentane are products of this reaction. Increasing the concentration of tributyltin hydride decreases the relative amount of methylcyclopentane, and decreasing the concentration of this hydride causes a linear increase in the percentage of methylcyclopentane (see if you can explain why).
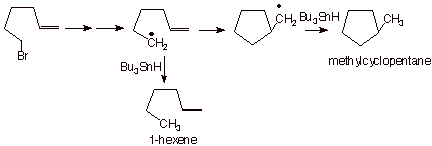
Scheme 13. Illustrating the Use of 6-Bromo-1-hexene as a Radical Probe.
Still another means of providing evidence to support the formulation of a radical mechanism is a stereochemical probe. This concept can be illustrated nicely in the reduction of organomercurials by sodium borohydride, a reaction which is popularly used in the context of oxymercuration/reduction of alkenes to afford net, anti-Markovnikov hydration of alkenes. The formulation of the reduction step as a (non-chain) radical reaction is supported by the reduction of exo- and endo-2-norbornylmercuric bromide by sodium borodeuteride. The observation that this substitution reaction is non-stereospecific is consistent with the intervention of a norbornyl radical, which can abstract a hydrogen atom from the exo face or the endo fact. The circumstance that the endo/exo product ratio is the same irrespective of whether the starting material has the endo or exo configuration, is consistent with the involvement of a common (radical) intermediate in both reactions. A concerted reaction, which would be expected to proceed with retention of configuration at C2 of the norbornyl system, is clearly ruled out by these stereochemical results. Since (undeuterated) water was included in the reaction medium, an intermediate carbanion should have been protonated, rather than deuterated. Finally, the 2-norbornyl carbocation is well known to react only from the exo fac, to give the exo-2-norbornyl product.
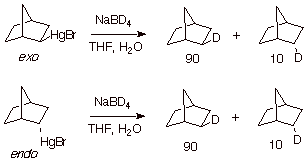
Scheme 14. Use of a Stereochemical Probe in the Reduction of An Organomercurial by Sodium Borohydride (Borodeuteride).
The detailed reaction mechanism is formulated in Scheme 15. Incidentally, the norbornyl cation reacts only from the exo face because it is a bridged (nonclassical) carbocation.
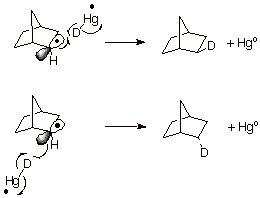
Scheme 15. Hydrogen Abstraction by the Norbornyl Radical from both the Exo and Endo Faces of the Radical.
Polar Effects in Radical Reactions
As is well known, radical chain bromination is highly selective for tertiary, allylic, or benzylic C-H bonds, as opposed to the relatively stronger primary or secondary C-H bonds. The bromination of toluene, for example, gives benzyl bromide. You should be able to write the radical chain mechanism for this reaction from your previous experience in organic chemistry. When the relative rates of bromination of m- and p- substituted toluenes are determined, a rather nice Hammett-Brown plot is found with r = -1.36. The negative value of rho indicates that a partial positive charge is being developed on the toluene moiety in the TS for the hydrogen abstraction step. The correlation with s+ rather than s indicates that the positive charge is being developed at the benzylic position (or less likely, directly upon the benzene ring). This is not necessarily surprising, since bromine atoms are relatively electronegative, and could draw electron density from the benzylic carbon in the TS (Scheme 16). What is, in a sense, rather unexpected, is that the correlation with polar substituent constants is so good (not perfect, but quite good). In other words, theTS has some carbocation character, but it also has extensive radical character, and the substitutents upon the benzene ring should have different abilities to stabilize radical character than carbocation character. Evidently, the
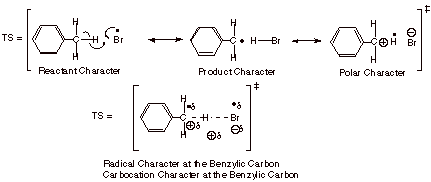
Scheme 16. Polar Substituent Effects Dominate Radical Stabilizing Effects in the TS for Hydrogen Abstraction from Toluene and Substituted Toluenes.
substituents’ abilities to stabilize radical character are much smaller in magnitude than their abilities to stabilize (or destabilie) carbocation character. As a consequence, the relative rates correlate rather well with polar substitutent consants (i.e., Hammet or Hammett-Brown parameters) and are but little affected by their (very different) abilities to stabilize radical character.
Radical Substituent Constants
So far we have not talked about substituent constants for purely radical stabilization effects. We understand that alkyl groups stabilize a radical center, so they should also stabilize radical character. We have also seen that groups which have unshared electron pairs, like methoxy, stabilize radicals by means of three electron bonding. This should also apply to such other substitutents as amino and halogen substituents. But, radicals are also stabilized by conjugation effects, so any groups which contain double or triple bonds in conjugation with the benzylic system (at the para position, in particular) should also be radical stabilizing. In fact, it is difficult to think of a substitutent which would destabilize a radical center. Consequently, it would be of interest to define a set of radical substituent constants. This turns out not to be as easy as one might think for the very reason that most radical reactions have TS’s which also have polar character, as in the case of the bromination of toluene, and in such cases the polar effect usually dominates the radical substituent effect. The desired set of substituent parameters has now been derived in various ways. One such approach is shown in Scheme 17.
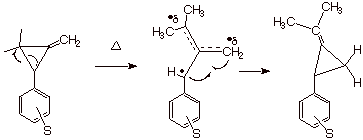
Scheme 17. Determination of Radical Substituent Constants from the Rates of Thermolytic Rearrangement. The TS, which is the Diradical Intermediate, Has Little, if any, Polar Character.
Some examples of radical substituent constants are given in the table below. Note that those given are for para substituents and that all substitutents are stabilizing relative to hydrogen. Recall that benzylic radicals have no radical character at the meta position, so the effects of meta substitutents upon the rate are small and involve indirect effects. Interestingly, most meta groups are mildly destabilizing. The relatively strong radical stabilizing effect of the dimethylamino substituent is noteworthy, and is a result of three electron bonding. The much greater ability of the methylthio substituent to stabilize a radical site than the methoxy substituent is also impressive. In the area of polar substituent effects, the methoxy group is much more effective at stabilizing a carbocation site than is the methylthio group. A similar circumstance applies in the comparison of a bromo and an iodo substitutent. It is likely that the enhanced effects associated with the heavier atoms derives from the ability to delocalize the odd electron via the 3d AO’s of sulfur and the 5d AO’s of iodine.
|
Para Substituent |
Sigma Dot |
|
NMe2 |
0.90 |
|
Vinyl |
0.67 |
|
NO2 |
0.57 |
|
Phenyl |
0.46 |
|
CN |
0.46 |
|
SMe |
0.43 |
|
I |
0.41 |
|
CO2Me |
0.35 |
|
MeO |
0.24 |
|
Br |
0.13 |
|
CH3 |
0.11 |
|
H |
0.00 |
Table of Radical Substituent Constants.
The Enhancement of Radical Chain Reactivity by Polar
Effects.
Austoxidation. It is well known that ethers should not be distilled to dryness because they often contain appreciable amounts of peroxides which, in the solid state, are potentially quite explosive. This is especially true of ether bottles which have been opened and exposed to air one or more times, because even at room temperature in an atmosphere which is only 20% dioxygen they are highly susceptible to oxidation. The radical chain mechanism for this so-called autoxidation is given in Scheme 18 . It is noted that hydrocarbons with no oxygen functionality are not reactive at all under these conditions. The especial sensitivity of ethers to autoxidation derives from the circumstance that the transition state for the hydrogen abstraction step is especially well stabilized by both polar and radical stabilizing effects. As shown in the scheme, the radical character is stabilized by 3 electron bonding, and the carbocation (polar) character is stabilized very powerfully by the unshared pair on oxygen (this is 2 electron bonding and is quite strong).
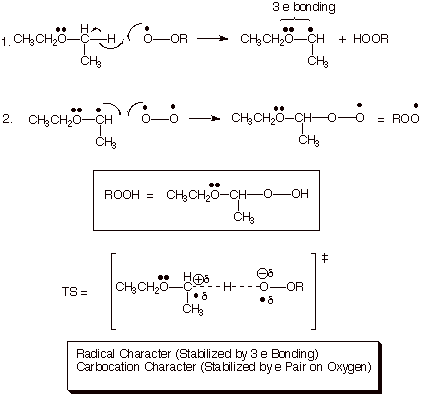
Scheme 18. Radical Chain Mechanism for the Facile Autoxidation of Ethers Selectively at the Alpha Carbon Under Extremely Mild Conditions. Both the Radical Character and the Carbocation Character in the TS are of an Exceptionally Stable Type.
We may note that the same is true of aldehydes, which are oxidized to peroxycarboxylic acids (Scheme 19). As an exercise, describe the type of radical stabilizing and carbocation stabilizing effects present in the transition state for the hydrogen abstraction reaction of an aldehydic hydrogen.