Unit 6: Anion Radicals
Addition of a single electron to a neutral (uncharged)
molecule generates a unique chemical species, called an anion radical (or radical anion by some) that simultaneously has a
unit of negative charge and an unpaired electron. This can be accomplished, in
many cases, by simply treatment with an alkali metal (which makes the alkali
metal cation the counterion to the anion radical). In general, the electron
will prefer (Aufbau principle) to enter the lowest energy antibonding molecular
orbital (LUMO). In the context of the resulting anion radical, this same MO
then becomes the SOMO (singly
occupied MO of a radical species). The LUMO of the neutral molecule is
therefore uniquely important among the ABMO’s because the added electron
will prefer to occupy that MO and also because it becomes the SOMO of the anion
radical, which (more or less) exclusively controls the distribution of the odd
electron, and therefore the position of likely reactivity of the radical.
Further, assuming that we are dealing with a hydrocarbon system as the neutral
precursor, and that this hydrocarbon itself is nonpolar (has zero charge
accumulations at all carbons), the SOMO also uniquely controls the distribution
of negative charge. In these cases, the spin and charge are said to be
tightly coupled, and in essence the
spin and charge “travel together”, i.e., the spin and charge at
each carbon are equal in absolute magnitude. This interesting result obtains
because, in the assumed kind of nonpolar system, the filling of the BMO’s
evidently results in Qi = 0 at each position. Therefore any charge
and any spin density must arise from the population of the SOMO. Needless to
say, if a polar functionality such as a ketone function is present, the filling
of the BMO’s does result in non-zero charge accumulations at most atoms,
so that both the SOMO and the BMO’s contribute to the overall charge
densities. Nevertheless, the SOMO continues to control the spin distribution.
In such systems, the spin and charge are said to be loosely coupled. They are
not uncoupled, because the SOMO continues to contribute most of the charge
density and all of the spin density. You have already calculated, as part of
the very first problem set in Unit 1, the charge and spin densities in the butadiene
anion and cation radicals, so that you can actually verify these things. Please
note also that the spin and charge densities in the anion and cation radicals
are identical (except for sign, in the case of charge).
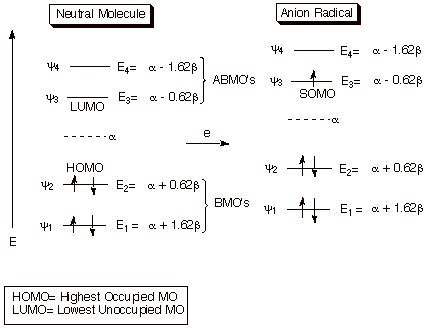
Scheme 1. Formation of an anion radical from a neutral
molecule by single electron acceptance.
Anion Radical Formation
It
should be noted that any organic molecule has antibonding MO’s, so that
essentially any molecule is capable of forming a corresponding anion radical.
When the LUMO is of very high energy, it may be difficult to transfer an
electron to the molecule by chemical means (such as an alkali metal) or even by
electrochemical means (cathodic reduction). Recall that pi bonds are weaker
than sigma bonds, in general. Thus, the energy of the pi BMO of ethene is not
as low as the sigma BMO of a C-C sigma bond. Since there is a pairing
relationship (with respect to the nonbonding level) between the BMO and the
corresponding ABMO, the pi ABMO of the ethene pi bond is not as high in energy
as the sigma ABMO of a C-C sigma bond (e.g. in ethane). Consequently, the
addition of an extra electron to a pi bond is much easier than to a sigma bond.
Nevertheless, the ABMO corresponding to the pi bond of ethene is still
relatively high in energy, so that experimentally, it is difficult to generate
the ethene anion radical. However, conjugation tends to decrease the energy of
the LUMO of a pi system (consider butadiene vs. ethene), and correspondingly
the addition of an electron becomes much easier to such conjugated systems
(Scheme 2). Finally, the insertion of an electronegative atom such as oxygen
has a strong energy lowering effect upon the LUMO, so that carbonyl pi bonds
are especially easy to reduce to anion radicals. In general, there is a very
nice linear relationship between the LUMO energy and the reduction potential of
the substrate.
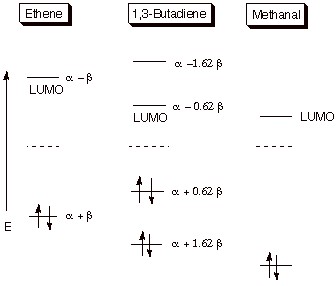
Scheme 2. Lowering of LUMO energies and raising of HOMO energies
via conjugation and incorporation of a heteroatom.
Examples of Stable
(Persistent) Anion Radicals.
If
sufficient delocalization and or heteroatom presence is provided, anion
radicals are not only relatively easily formed, but they can be stable enough
to persist in solution in the absence of proton donors or oxygen. The blue
benzophenone anion radical is very commonly seen in the organic laboratory as a
drying agent for ethereal solvents such as tetrahydrofuran (THF). The idea is
that small amounts of water which may be present in the solvent would quickly
protonate the basic anion radical. Atmospheric oxygen would also be removed by
reaction with the anion radical. Therefore if the blue color of the
benzophenone anion radical persists, the solutions can be considered to be very
dry and oxygen-free. The procedure for drying such a solvent is more or less as
follows. Starting with a flask of already rather pure and anhydrous solvent
(obtained commercially), a small
amount of benzophenone is added followed by the addition of potassium metal.
The initial amounts of the benzophenone anion radical will form and then
disappear as a result of reaction with water and/or oxygen. After a while, the
blue color will persist, showing that the solvent is now rigorously anhydrous
and anaerobic. Distillation of the solvent then yields a very pure sample. Note
that the benzophenone anion radical (ketyl) benefits not only from extended
conjugated (over both phenyl rings) but also from the electronegative
heteroatom effect.
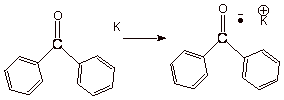
Scheme 3. The
benzophenone anion radical (ketyl): A persistent anion radical.
Another
very common anion radical seen in the organic lab is the biphenyl anion radical
(green). Usually this is prepared by reaction of biphenyl with lithium metal,
often in THF solvent. This anion radical, and other anion radicals of aromatic
systems, is often used as an electron transfer reductant. It is interesting to
note the relative stability of the anion radicals of aromatic system, even
though the addition of an electron to an ABMO diminishes the net bonding of the
system. Evidently, some of the special stability associated with the
aromaticity of the neutral substrate still remains in the anion radical.
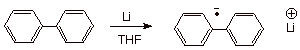
Scheme 4. The biphenyl anion radical: A one electron reducing
agent.
Stable Anion Radicals. The tetracyanoethylene anion radical has already been
mentioned as an example of an isolable anion radical salt. Another example is
the tetracyanoquinodimethane anion radical (Scheme 5).

Scheme 5. The TCNQ anion radical.
The Cyclooctatetraene
Anion Radical. The cyclooctatetraene
anion radical is a persistent anion radical in solution in the absence of air
and moisture (Scheme 5). It is especially interesting for a number of reasons.
First of all, its neutral precursor, cyclooctatetraene has a non-planar, tub
shape. This is not especially surprising in view of the fact that the planar
form would be a cyclic, conjugated system consisting of 8 pi electrons. It
would, accordingly, be classified as antiaromatic, according to the 4n+2 Rule.
The anion radical, however, is a 9 electron system and although not aromatic,
it is not antiaromatic either. As it turns out the anion radical of this system
is actually planar and a true cyclic, conjugated system.
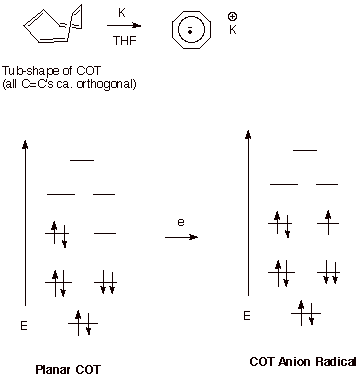
Scheme 6. The planar cyclooctatetraene anion radical.
Another
aspect of the COT anion radical is that it disproportionates to a somewhat
surprising extent to the corresponding dianion and a molecule of neutral
cyclooctatetraene (Scheme 6). The driving force for this can be readily seen,
since the dianion, as a 10 electron system, can be considered to be aromatic.
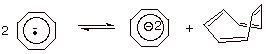
Scheme 7. Disproportionation of an anion radical.
Reactive Anion Radicals
The reduction of 1,3-butadiene by alkali metals
dissolved in liquid ammonia results in conjugate addition of hydrogen to the
diene system, i.e., formation of 2-butene (cis and trans isomers). As background, we should note that alkali metals
dissolved in liquid ammonia to give deep blue solutions of solvated electrons
in the very polar solvent, ammonia. These electrons are available to transfer
to an ABMO of an organic molecule. Although it is difficult to reduce ethene to
its anion radical, the lower energy LUMO of butadiene easily accepts an
electron to form the corresponding anion radical. Hopefully you have already
calculated the charge densities in
the 1,3-butadiene anion radical (first unit) and found that most of the
negative charge resides on C1 and C4 of the diene. It would not be a surprise,
therefore, to find that if protonation of this anion radical occurs, it will
occur on one of these terminal atoms. The result is the formation of an allylic
type radical. Since the SOMO in the allyl radical is an NBMO, this radical is
easily reduced (by more solvated electrons) to the corresponding allylic anion.
This anion is then protonated (by ammonia or an alcohol which has been added to
the solvent) to yield 2-butene. This protonation occurs rather selectively on
the terminal atom to yield 2- rather than 1-butene. The mechanism of this
reduction, which is typical of the reduction of conjugated dienes (or enones)
by metals in liquid ammonia, is shown in Scheme 7.
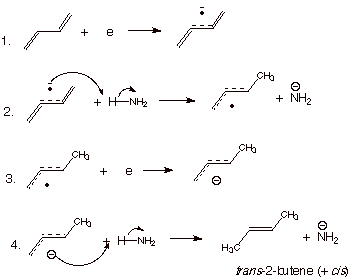
Scheme 8. Reduction of a conjugated diene with alkali metal in
liquid ammonia.
In
a similar way, naphthalene is reduced to the specific dihydroisomer shown in
Scheme 9. Incidentally, the position of protonation of an anion radical of an
alternant hydrocarbon system can be confidently predicted by a knowledge of the
SOMO distribution, since this MO exclusively controls both the spin and the
charge in these systems. In the naphthalene anion radical, the SOMO has the
largest coefficients on the alpha positions, so that initial protonation occurs
there. Reduction of the resulting radical to an anion is followed by
protonation, also at the alpha position.
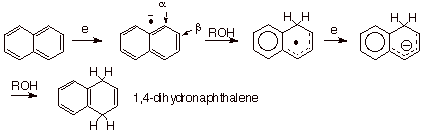
Scheme 9. Birch reduction of naphthalene by sodium in liquid
ammonia, containing tert-butyl
alcohol.
Incidentally, even benzene can be reduced to 1,4-dihydrobenzene in this way. These reactions represent examples of Birch reduction.
The Pinacol Coupling Reaction
The reductive coupling of two ketone molecules by
magnesium metal to give a 1,2-diol, often termed a pinacol coupling, is
considered to take place via the anion radical of the ketone (recall that the
anion radical of a ketone is called a “ketyl”). We should pause
here to consider a few special aspects of ketyl anion radicals. First of all,
the ketone carbonyl pi bond is highly polarized, with a large fractional
negative charge on oxygen and a corresponding positive charge on carbon. This
corresponds to a BMO which is more heavily weighted on oxygen than on carbon
(Scheme 10). Perhaps not too surprisingly then, the ABMO shifts over to having
a heavier weight on carbon. Since the ABMO will be the SOMO of the anion
radical, and since the SOMO controls the spin density distribution, the radical
character will be greater on carbon than on oxygen. Since, however, the neutral
ketone already had much negative charge on oxygen before being converted to the
anion radical, and the carbon had positive charge, the SOMO does not
exclusively control the charge distribution, i.e., the spin and charge are
uncoupled. The electron density which goes onto carbon from the SOMO is used in
part to neutralize the positive charge on carbon, and that which goes onto
oxygen further increases the net negative charge on oxygen. In effect then,
this ketyl has the radical character mostly on carbon and the charge density
mostly on oxygen. Radical coupling, then, occurs at the site of highest spin
density, i.e., carbon.
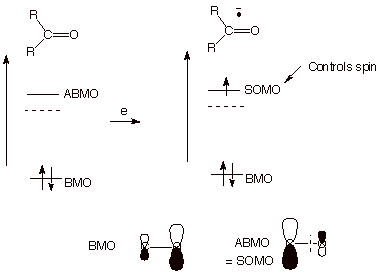
Scheme 10. Spin and Charge Uncoupling in a Ketyl
Since magnesium has two electrons outside of a closed shell, it is a good electron transfer donor (reducing agent). In a heterogeneous reaction, it transfers an electron to the ketone, giving a ketyl. Two of these ketyl anion radicals than couple to give the magnesium salt of the double conjugate acid of the pinacol (Scheme 11).
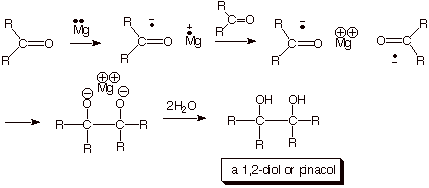
Scheme 11. Coupling of two ketyl anion radicals in the formation
of pinacols.
Anion Radical Cycloadditions
Here at UT, we have been developing some very novel anion radical cycloaddition chemistry. Recall that the formation of cyclobutanes by cycloaddition of two alkene molecules is extremely difficult and, for practical purposes, quite useless. However, it turns out that placing electrons in an antibonding MO raises the energy of the species sufficiently that such reactions are quite feasible in some cases. Intramolecular reactions such as that shown in Scheme 12 have been found to proceed quite efficiently under electrochemical reducing conditions or using persistent anion radicals of aromatic systems to generate the required anion radicals.
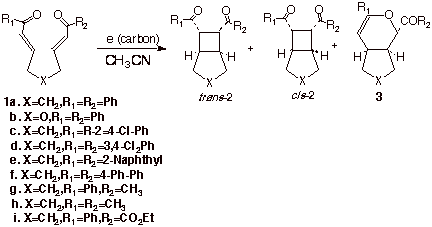
Scheme
12. Intramoolecular
Anion Radical Chain Cyclobutanation
The
mechanism proposed for these reactions is shown in Scheme 13.
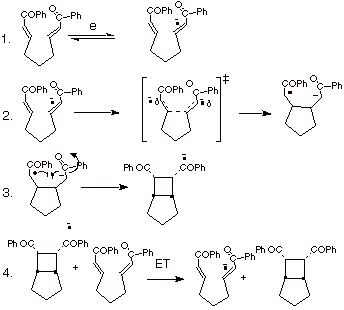
Scheme 13. Mechanism of Anion Radical Chain Cyclobutanation
Examples of intermolecular anion radical cyclobutanation have also been developed in this department and are currently under exploration.