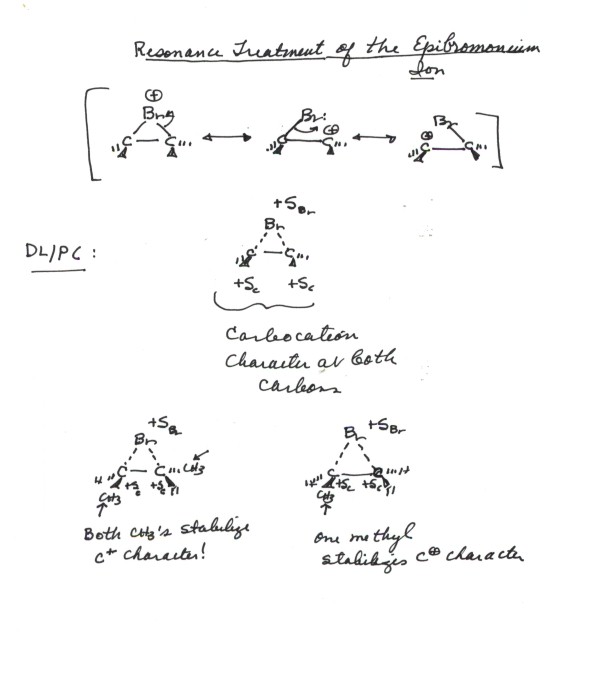
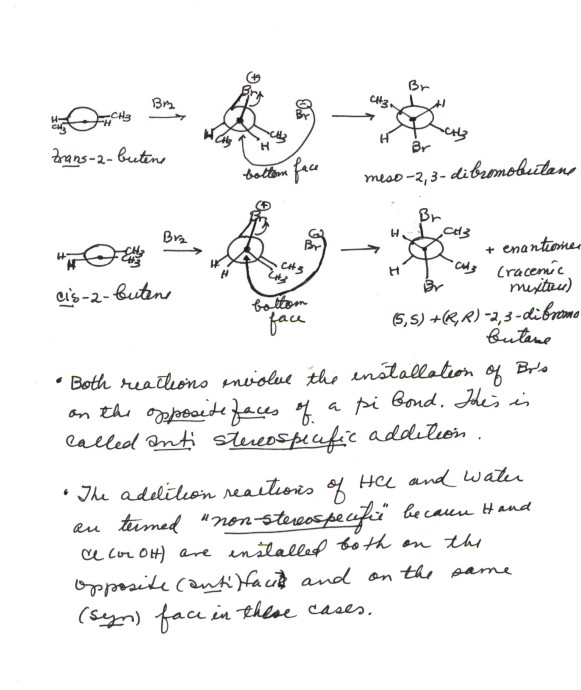
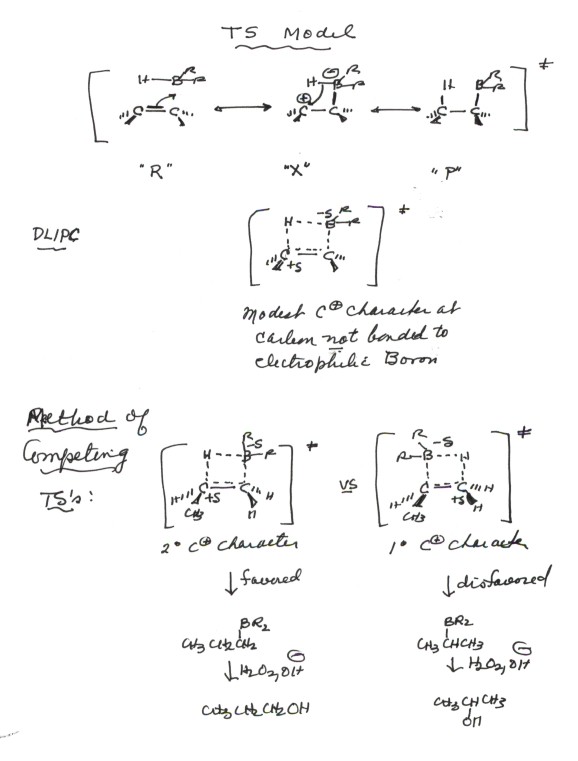
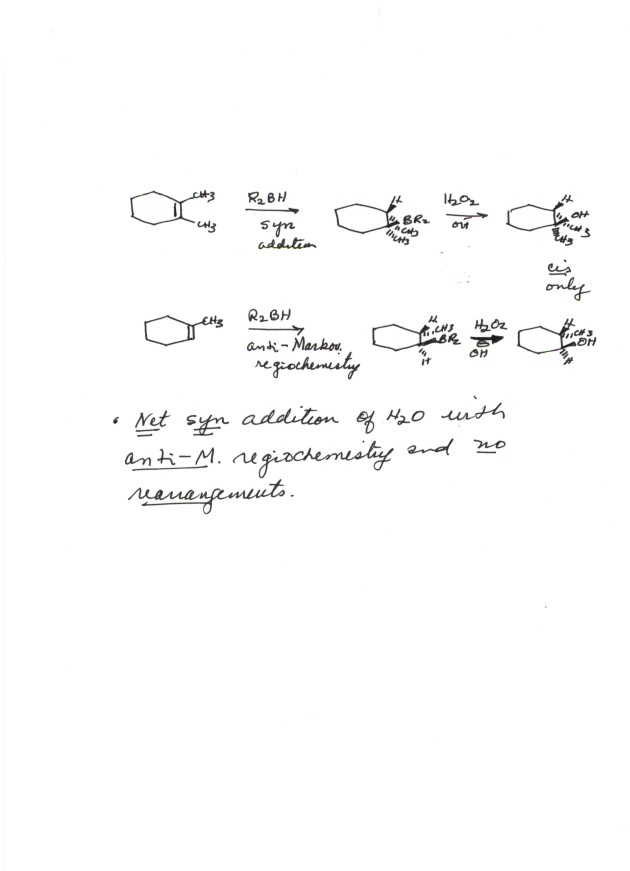
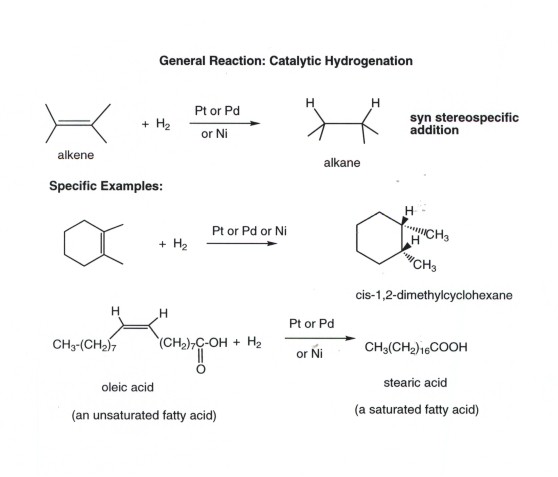
ADDITION OF WATER TO ALKENES (HYDRATION)
THE OVERALL REACTION
The acid catalyzed addition of water to a general alkene and also to isobutene is illustrated below:
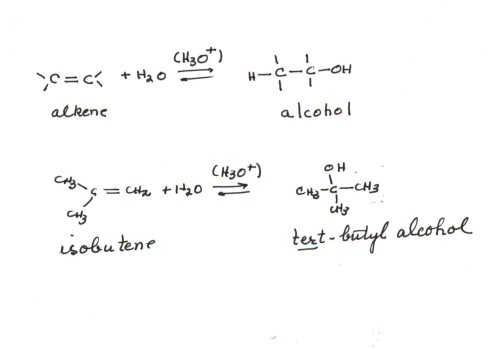
- Note that this is not a reaction mechanism, but an equation for the overall reaction.
- Hydronium ion is a required catalyst. Since it is not consumed in the reaction, it is not a part of the equation per se, but is placed in parentheses over the reaction arrow.
- Note also that the product is an alcohol.
- Note that the Markovnikov Rule is followed for this reaction, i.e., the proton adds to the less substituted carbon of the alkene double bond (the methylene carbon, making it a methyl group), and the hydroxyl group adds to the more highly substituted carbon atom of the alkene double bond. So the regiochemistry of the hydration reaction is closely parallel to that previously discussed for the hydrochlorination reaction.
MECHANISM OF ACID CATALYZED HYDRATION

- In the addition of HCl, the acid which transfers the proton to the alkene to form a carbocation is, of course, HCl. In acid catalyzed addition of water (hydration), the acid which actually transfers the proton to the alkene is normally the solvated hydronium ion. The acid used is often sulfuric acid, but any strong acid will usually work. Water, itself, is too weak an acid to transfer the proton to the alkene, so acid catalysis is essential.
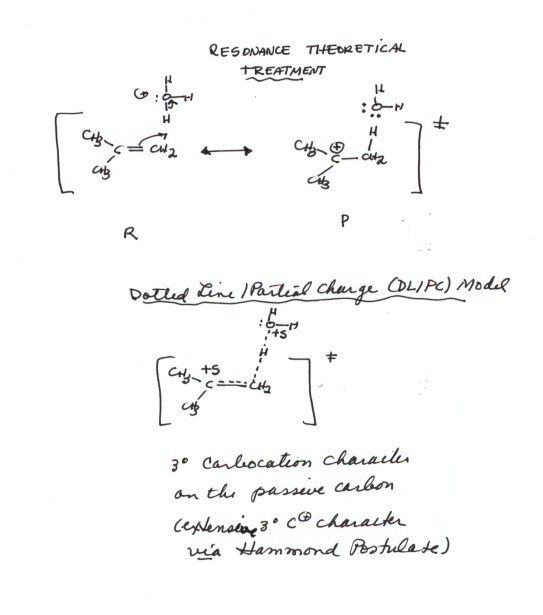
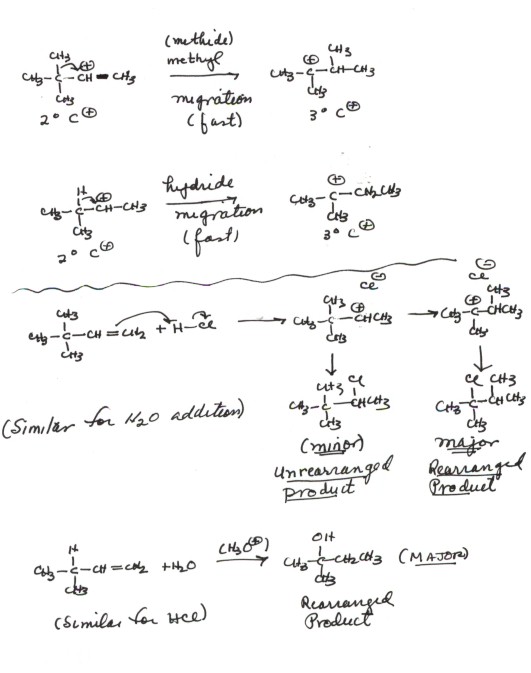
- The reaction is also similar to the addition of HCl in that a carbocation is formed in the rds. Again, two bonds are broken and just one is formed, so the reaction is endothermic. It is therefore the rds, and the TS strongly resembles the product carbocation (Hammond Principle). However, it differs in that the carbocations, which are very reactive and so can react even with a weak nucleophile like water, which is present in great abundance, react with water to give the conjugate acid of the product alcohol.
- In the final step of the mechanism , this conjugate acid transfers a proton to water, regenerating the original hydronium ion catalyst. It is important to note that this step is an equilibrium step, since proton transfers from oxygen to oxygen are typically very fast. IT IS VERY IMPORTANT, IN WRITING A CORRECT REACTION MECHANISM, TO SPECIFY BY THE USE OF AN EQUILIBRIUM ARROW ANY STEP WHICH IS REVERSIBLE. IT IS ALSO IMPORTANT TO SPECIFY THE RDS. FURTHER, YOU MUST USE ELECTRON FLOW ARROWS TO SHOW THE FORMATION OF NEW BONDS. The product alcohol would be isolated by making the solution basic or by extracting it into an organic solvent or distilling it off.
TRANSITION STATE MODEL FOR HYDRATION
The development of a TS model for the rds of hydration using resonance theory is illustrated below. You should be able to show this same kind of development for any alkene.