ALKENES II: REACTIONS OF ALKENES
TOP
TABLE OF CONTENTS FOR THIS
PAGE
- Reaction types
- Reaction Mechanisms
- Transition State Theory
- Reaction Path Diagrams
- Reaction Rates and Rate Constants
- An Example of a Reaction Mechanism
- Reaction Path Diagram For this Example
- Transition States
- Transition State Models
- Resonance Theory of Transition States
- Regiospecificity and Competing Transition
States
- Relative Rates of Addition to Different
Alkenes
- Stabilization of Carbocations by
Alkyl Groups
- Transition State Refinement: The Hammond
Postulate
REACTION TYPES
- Organic reactions are classified as to Reaction Type
based upon the overall changes which occur in the product as compared to the
reactant(s).
- For alkenes the distinctive reaction type is Addition.
- This is because the pi bond of an alkene can be broken and
this bond replaced by bonds to other (external) agents. For example, ethene
can add molecular hydrogen to give ethane. In this reaction, two reagents
add together to give a single product. This is typical of the type
of reactions that alkenes undergo.
- Alkenes are especially reactive because the pi
bond is relatively much weaker than a sigma bond.
- In contrast, alkanes cannot undero addition at all,
because the carbons of an alkane already have the maximum number of atoms
(four) bonded to them. They therefore cannot add another atom. For example
ethane can not add more hydrogen . Substitution,
where, for example, a hydrogen atom of an alkane is replaced by some other
atom or group, such as a halogen atom is the typical reaction.
- The symbol for an addition reaction is "Ad",
wherease that for a substitution is "S".
REACTION MECHANISMS
- A Reaction Mechanism is a stepwise description
of the reaction path. In our course this will consist of a set of chemical
equations along with certain other specifications (to be specified later)
such as the rate determining step.
- The term Reaction Path refers to the lowest
energy pathway from the reactants to the products, i.e., the one
which is actually preferred and followed in a chemical reaction. Many
paths are conceivable, but the easiest and fastest way to convert reactants
to products is via the lowest energy path.
- The importance of knowing the mechanism of a reaction lies
in the ability to (1)exert some control over the reaction in regard to its
rate and yield, and in general its efficiency and (2) the ability to predict
the precise stereochemistry and other subtle aspects of the structure of the
product.
- In our discussion of reaction mechanisms, we will designate
a general reactant or group of reactants as "R" and the product
of this general reaction as "P".
- Some reactions occur in a single elementary reaction
step. That is, all of the bonds which are to be broken in the reactant
and all of the bonds which are to be formed in the product are broken and
formed in one step. Such reactions are called "Concerted Reactions".
- Most reactions occur in two or more steps (possibly many).
Such reactions are called "Stepwise Reactions". In a stepwise
reaction, the formation of intermediates (to be defined) is involved.
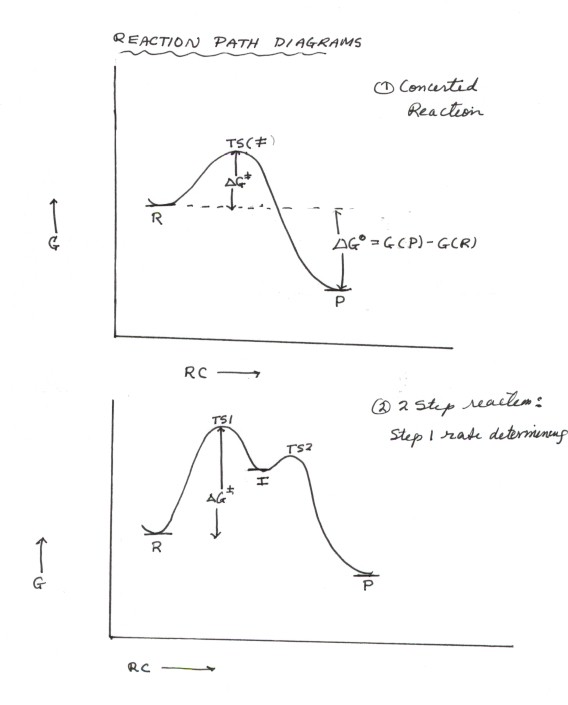
TRANSITION STATE THEORY
- Consider a simple, one-step reaction. The reactant R is
converted to the product P by breaking one or more bonds of R and forming
one or more new bonds as found in P. The process of breaking bonds thus
requires energy, so nearly all reactions require some input of energy,
called activation energy.
- This can be illustrated by considering what we call a
Reaction Path Diagram, which plots the energy
(usually in uncalibrated units) on the vertical axis versus the Reaction
Coordinate, which is a measure of the progress of the reaction
from the geometry of R toward that of P. The point of highest energy on
this reaction path is called a Transition State.
- Formally, a transition state is defined as the state
of highest energy on the lowest energy reaction path. An important consequence
of this definition of the TS is that the energy difference between the
transition state (abbreviated as TS or as a double dagger) and that of the
reactant(s) is the activation energy. We will see
that this activation energy controls the rate of the reaction: the
lower the activation energy, the faster the reaction goes, and the higher
the activation energy, the slower the reaction goes.
- Various energy parameters can be used on the vertical axis.
We will start out by using the quantity G, the Gibbs free energy, because
rigorously this is the quantity which actually controls the rate. Other options
are the enthalpy H or the chemical energy,E, which are less directly related
to the rate.
- It is important to note that the (1)the energy difference
between the R and P controls the thermodynamic equilibrium between R and P
and that (2)the energy difference between the TS and R controls the rate
of the reaction. An important consequence of this is that the energy
of the product does not directly enter into the quantity (activation free
energy) which controls the rate of a reaction. However, the TS energy
obviously cannot be less than that of the P.
- In a two step reaction, illustrated in the second diagram
below,there is an intermediate state which, like the reactant and product,
is an energy minimum. This state is designated as I on the reaction path diagram
. The formal definition of an intermediate is an energy minimum encountered
on the reaction path between R and P . The physical significance of being
an intermediate, as opposed to a TS, is that a substance which exists at an
energy minimum has a significant lifetime and, in many cases, its presence
can be physically detected and its reactions chemically diverted if desired
to another course. A TS is so short lived that it cannot be detected by conventional
physical means, and its chemistry cannot be affected by any other reagent.
- In a two step reaction, there are two extreme scenarios,
i.e., either the first step is rate determining (we call such a step the rate
determining step or rds) or the second step is rate determining.(By extreme
we mean that possibly neither step will be rigorously rate determining). The
scenario illustrated below is one of the former type in which the first step
is rate determining. The concept of a rds is extremely important in dealing
with reaction rates of organic reactions. It is defined as a reaction step
the rate of which is equal to the overall rate of the reaction.
The importance of the concept is this: If the rate of a single step is exactly
equal to the overall rate of product formation (or of reactant consumption,
which should be the same thing), anything we know about the rate of this step
will immediately help us to know about the rate of the overall reaction. Thus,
if a reaction has 10 steps and one of them is rate determining, we really
don't need to know anything about the rate of 9 of them, if all we are interested
in is the rate of the overall reaction. Conversely, anything we learn about
the overall reaction rate immediately tells us about the rds and it alone.
- The reasons why the first step in the illlustrated diagram
is the rds are twofold: (1)Once the intermediate I is formed, it virtually
always goes on to product, as opposed to returning to the starting materials
(reactants). Why is this? Simply look at the height of the two barriers separating
I from P and from R. Note that the barrier for return to R is substantially
higher than that separating I from P. So the rate of return to R is much slower
than the rate of conversion to P. (2) The second requirement is that the
concentration of the intermediate must not build up, i.e., I is very quickly
converted to P, so that there is very little hold up. Thus, the overall picture
is that I essentially never returns to R and it doesn't buld up, but goes
quickly over to P .So, as fast as I is formed it is conerted to P. That makes
the rate of formation of I essentially equal (for all practical purposes,
equal) to the rate of formation of P.
- If the second step of the reaction is rate determining,
this corresponds to the first step being an equilibrium. That is the intermediate
rapidly goes back to R and only slowly on to P. In such a case, the activation
energy for the reaction is the energy difference between the transition state
of higher energy (in this case the second TS) and the R. See if you can
draw a reaction path diagram which proprerly shows that I goes back to R faster
than it goes on to P.
REACTION PATH DIAGRAMS
Reaction Rates and Rate Constants
Consider an elementary reaction step such as the one shown
below:

- The rate of the reaction is the rate at which P is formed
or R is consumed. This can normally be measured experimentally by simply
analyzing the amount of R present at various times.
- The rate is proportional to the concentration of R to the
first power, since the more R we have, the faster P is formed.
- The proportionality constant, designated as k (small
k, to distinguish it from the equilibrium constant K), is called the rate
constant. We should make a very clear distinction between the rate of a
reaction (the overall rate) and its rate constant. The rate constant
is a measure of the reaction rate at unit concentration of the reactant,
so that it is a measure of the inherent speed of the reaction, and is independent
of the concentration of the reagent. Any reaction can be speeded up by increasing
concentrations of reagents, but this doesn't affect the rate constant.
- It is this measure of the inherent rate of a reaction which
is related directly to the free energy of activation. The larger the activation
free energy required, the smaller the rate constant.
- Again, if a given step of a multistep reaction is the rds,
the rate of the overall reaction giving the final product is still equal to
the rate of this step. So the rate law for this single step gives the rate
law for the overall reaction.
- For any reaction, be it a single or multi-step reaction,
the expression which gives the overall rate of the reaction in terms of rate
constants and concentrations of reagents is called the Rate Law.
An Example of a Reaction Mechanism: Addition
of HCl to An Alkene
CONSIDER THE ADDITION OF HCl
TO ISOBUTENE
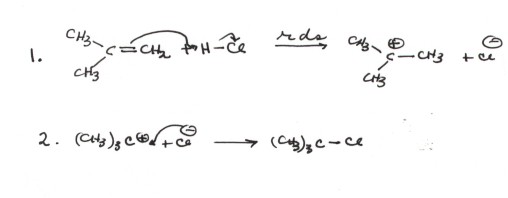
IMPORTANT NOTES ABOUT THIS
MECHANISM
- First, please note the format and conventions of writing
mechanisms. You must always write the different steps of a mechanism on
separate, numbered lines.
- You must always designate, if known, the rds. Also, if
any step is an equilibrium, this must be indicated by the use of equilibrium
arrows.
- You must show by means of arrows the flow of electrons
into the new bonds. In the first step, the electron pair of the pi bond
flows toward the hydrogen of HCl to form a carbon-hydrogen bond to one of
the alkene carbons, the other being left then with a positive charge (as a
carbocation). Since hydrogen cannot be divalent, the electron pair of the
H-Cl bond must flow onto the chlorine atom, generating a chloride anion. In
the second step, the electron pair on the chloride anion flows toward the
carbocation center to form a C-Cl bond.
- In this reaction, the overall result is the addition of
HCl to isobutene. The tert-butyl carbocation is an intermediate. Being
very reactive, it doesn't build up, but always goes on to the product. The
first step is the rds, while the second step is fast and not reversed.
- You should be able to explain why the first step is the
rds.This is really quite simple. The second step breaks no bonds and forms
a bond. It is therefore very favorable energetically ( it has a negative delta
G) and because no bonds are broken, it is very fast. However, in the first
step two bonds must be broken, and only one is formed. The breaking of two
bonds is in itself difficult, and in this case the reaction is also energetically
very uphill (unfavorable) because only one new bond is formed in the first
step. In general, with reasoning like this (simply noting how many bonds are
broken and formed), you can adequately rationalize or even predict which step
of a reaction will be the rds.
- Finally, please note that the carbocation intermediate is
very reactive because in it the carbocation center is only trivalent and it
can therefore readily form a new bond without breaking any bonds. It is
a strong Lewis acid.
A REACTION PATH DIAGRAM FOR
THIS REACTION
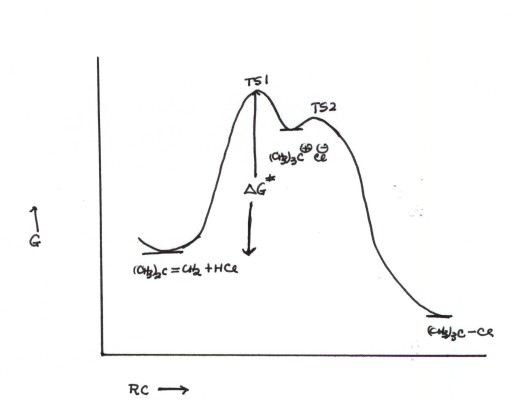
THINGS TO NOTE ABOUT THIS RP
DIAGRAM
- Note that, overall in this reaction, two bonds are formed
(C-H and C-Cl) and two are broken (the C-C pi bond and the H-Cl bond), but
these are not all broken and formed at once. The lowest energy reaction path
involves breaking two of these bonds and forming one in the first step.
- Again, note that an intermediate is an energy minimum. This
is the tert-butyl carbocation/chloride ion pair. Note that in the drawing,
the barrier to going on to the product (second step, via TS2) is smaller than
the barrier to going back to the reactant (via TS1). So the intermediate always
goes on to product , and the first step is rate determining.
- Note that the activation free energy is that of the first
step, since it is rate determining. No further activation is required, since
the intermediate always goes on to prodcuct, and never builds up in concentration.
- So, although there are two steps and two TS's, the second
one is of no consequence as far as the rate is concerned
TRANSITION STATES AND TS MODELS
THE NATURE AND IMPORTANCE OF TRANSITION STATES
- Transition states have extremely short lifetimes (less than
about 10 -13 seconds), so they are extremely difficult to detect.
We therefore have very little experimental knowledge about them. They exist
for so short a time (usually the time required for the vibration of a bond)
because they are at the top of the energy curve and require no further activation
to go on to product.
- Nevertheless, TS's are extrememly important, especially
in the context of reaction rates (they are irrelevant in the context of
equilibria; why?) because the energy of the TS relative to the reactant
determines the energy barrier, which in turn determines the rate of the reaction.
- Therefore, it is important that we have some information
about them, even qualitative information, in order to organize our knowledge
about the reaction and especially to be able to make some predictions about
relative rates.
Transition State Models
- For the above reasons,it is extremely valuable to have even
a rough and qualitative structural model of the transition state of a reaction,
such as the addition of HCl to isobutene. Such a model would contain information
about the magnitude and location of any charge which is generated in
the TS, an indication of any bonds which are being made or broken,and
other things.
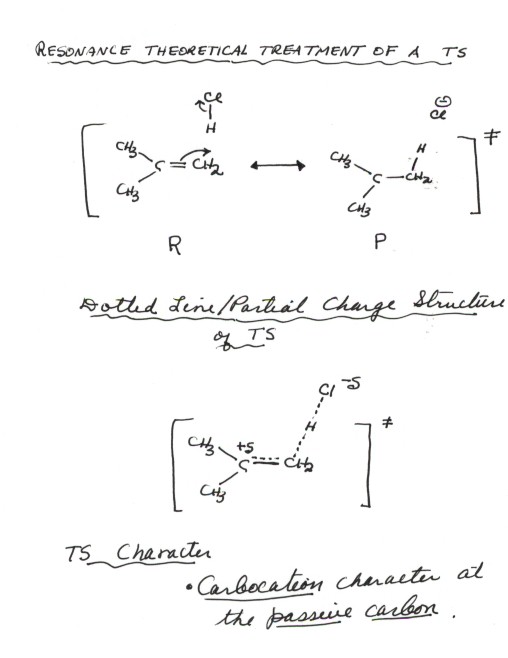
- Resonance theory,
which we have discussed previously, provides a convenient way to derive a
TS model for any reaction. Since a TS is intermediate in geometry between
the reactant and product, it should be possible to represent it, using resonance
theory, as a resonance hybrid of reactant and product-like structures.
In fact, these should normally be the best two canonical structures. It may
be necessary, later,to provide additional structures also, but any transition
state should be a least reasonably well represented as a hybrid of these two
main structures.
- In applying resonance theory, it is important to recall
that all resonance structures apply to a common geometry, in this case,
that of the real TS, even though this may not be precisely known. The important
factor here is the two reactants in our case must be oriented at least
roughly as we anticipate them to be in the TS. In other words (see the
resonance structures below) with the H end of the H-Cl molecule approaching
a single carbon of the alkene double bond, since the proton is obviously going
to bond to one carbon.
- In the case of the first step of the reaction of H-Cl with
isobutene, which is the rds, the reactant-like (or R-like) structure is that
of isobutene and HCl, properly oriented. The product-like (or P-like) structure
is that of the tert-butyl carbocation/chloride ion pair, again properly
oriented. If we write the R-like structure properly oriented, we can generate
the P-like structure just by electron flow arrows, as shown. These electron
flow arrows should be shown when you are using TS theory to derive a TS model.
- Dotted line/partial charge (DL/PC) Structures. This
completes the first step in deriving a TS model for our reaction step. We
can make this easier to analyze by summarizing the resonance theoretical treatment
in terms of a dotted line/partial charge (DL/PC) model. This is done
simply by representing any bonds which are being made or formed (i.e., any
bond which is made in the R and broken in P, or conversely) as dashed lines.
These dashed lines imply a Partial Bond. Secondly, partial charge accumulations
are represented by +d or -d
(lower case Greek delta) at the proper location. Any position which is
charged in one structure and not in the other has a partial charge.
- Our DL/PC model of the rds of the HCl/isobutene reaction
should now show the C-C pi bond as partial, as well as the H-Cl bond, both
of which are being broken. It should also show as partial the C-H bond which
is being formed. A partial positive charge is on the carbon which is not bonding
to the proton. We will call this the passive carbon since it is not
being attacked by the proton.
- We will see momentarily how this very simple TS model can
aid us in thinking about the results of organic reactions, not only reaction
rates but products of reactions.
- Finally, it wil be important for us to know how to think
with these TS models. What aspects or characteristics of the model are likely
to yield important insights into the reaction. We call this "Characterizing
the TS". In this case, the main character of importance in the TS
is THE DEVELOPMENT OF CARBOCATION CHARACTER AT THE PASSIVE
CARBON. Note that the character and its location are both specified.
Later, we will learn to further refine and extend this TS character.
RESONANCE THEORETICAL TREATMENT
OF A TS
REGIOSPECIFICITY AND TS MODELS
- Regiospecificity: Let's
begin immediately to put the TS model and its character to use. We can do
this by considering the regiochemistry of the HCl addition reaction.
Note (see illustration) that there are two senses in which HCl could potentially
add to isobutene, one which gives the product actually observed, tert-butyl
chloride and one which would have led to isobutyl chloride. Essentially,this
corresponds to a very high degree of selectivity for the proton adding to
the CH2 end of the alkene and the Cl adding to the other end, as
opposed to the opposite situation. This orientational selectivity is called
"regioselectivity" or "regiospecificity"
(organic chemists can't seem to make up their minds about this) . Of course,
this does not even come up in the case of a symmetrically substituted alkene
like ethene, or even with isobutene when it reacts with a symmetrical reagent
like Cl2. It only comes into play when two unsymmetrical reagents
react.
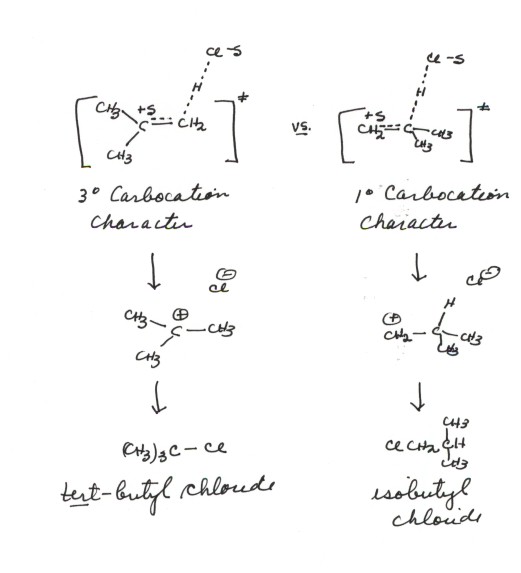
- Method of Competing Transition States: Let's
now attempt to represent a TS model for each of these two reactions. Since
we have already developed the model for the observed isobutene reaction, we
don't have to go all the way back and do the same thing for the other regiochemical
sense. All we have to do is turn the isobutene around so that the proton is
bonding to the more hindered end of the alkene (illustration).The TS models
are very similar, except that the carbocation character is being developed
in one case at a tertiary carbon and in the other case at a primary carbon.
We will thus refine our TS characterization to read "tertiary carbocation
character at the passive carbon" in one case, and " primary
carbocation character at the passive carbon' in the other.
- In a short while , we will learn that tertiary carbocations
are much more stable than primary carbocations(and why). Thus, the TS
with tertiary carbocation caharacter is much more favorable (lower in energy)
than the TS with primary carbocation character. We can thus see why the reaction
proceeding via the tertiary TS is much faster, and why the tert-butyl
chloride product is essentially the only one observed.
,
TS MODELS RATIONALIZE THE OBSERVED
REGIOCHEMISTRY
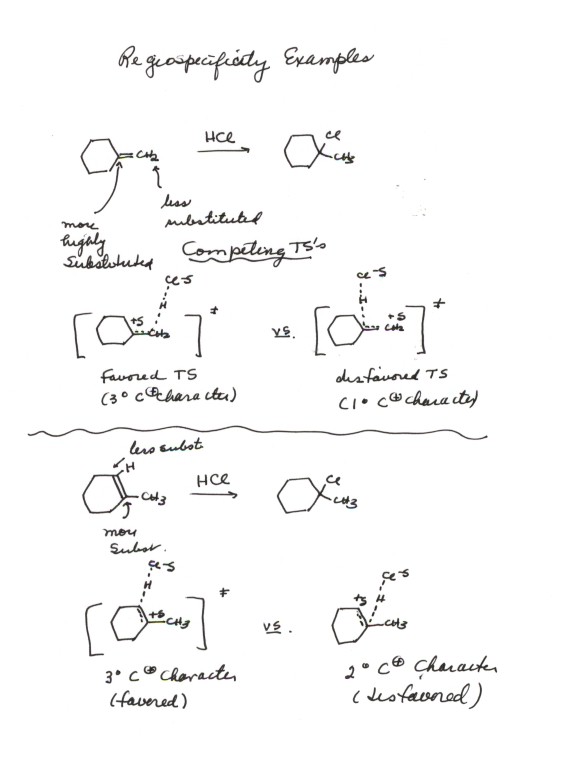
- The general procedure we will use for such comparisons is
the METHOD OF COMPETING TRANSITION STATES.
We, of course, use TS Models of the TS's of the competing reactions. The lower
the energy of a series of competing transition states, the lower the activation
energy, and the faster the reaction. The reaction which will be preferred
is the faster one, of course.
- In the diagram above,the TS which has tertiary carbocation
character is of lower energy than that which has primary carbocation
character, because positive charge on a tertiary carbon is more stable than
ion a secondary or primary carbon. The lower the energy of the TS, the lower
the activation energy and the faster the reaction. Thus protonation of the
methylene carbon is faster than protonation of the carbon bearing two methyl
groups. [Incidentally, there is also a steric factor which favors the approach
to the less sterically hindered methylene carbon]
- In general, the development of tertiary carbocation
character in a TS is more favorable than secondary carbocation character,
which is more favorable than primary carbocation character. The least
favorable is methyl carbocation character.
- The product of the reaction of HCl with isobutene is thus
tert-butyl chloride (i.e., 2-chloro-2-methylpropane) and essentially
no isobutyl chloride (i.e., 1-chloro-2-methylpropane) is formed. In the case
of addition to propene, isopropyl chloride is formed, and no substantial amount
of 1-chloropropane (secondary vs. primary carbocation character in this case).
The regiochemistry of addition is predicted by a qualitative rule, called
the MARKOVNIKOV RULE: : "In electrophilic additions of HX, the
proton adds to the less highly substituted carbon atom of the
double bond". We can now understand , and we should be able to explain,
that this is a result of the development of positive charge on the more highly
substituted carbon atom, where the charge is more highly stabilized. (Recall
that carbocation character is developed not on the carbon being attacked by
the proton, but on the "passive carbon".
- Two other additional examples are provided in the diagram
below.You should be able to predict the orientation of the addition of
HCl to virtually any alkene using (1)the Markovnikov Rule and (2)rationalize
it bY drawing competing transition states for the two modes of addition.
RELATIVE RATES OF HCl ADDITION
RELATIVE RATES OF ADDITION
TO DIFFERENT ALKENES
- The relative rates of addition of HCl to different alkenes
can also be predicted using the same Method of Competing TS's. This
is important tool, because the rates of addition to some alkenes may be slow,
to others modest, and to others fast. The alkenes which react very slowly
may need to be reacted at a higher temperature in order to obtain a reasonable
reaction rate. We need to know which alkene we are dealing with and whether
to expect its reaction to be fast or slow. This can be especially important
when two or more alkene functions are present in the same molecule. We
could then predict which one of them will react fastest, and may be able to
leave the less reactive one intact.
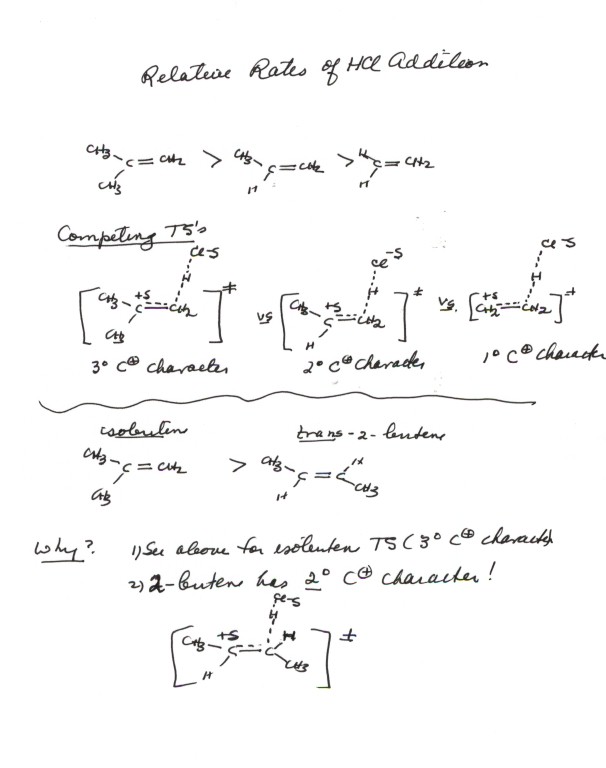
- In the reaction series isobutene, propene, and ethene(shown
above) the first named alkene reacts very fast, while the last one reacts
relatively slowly. Propene reacts at an intermediate rate. Such a sequence
of relative rates can be easily predicted or explained, by observing that
the TS's for the additions have tertiary, secondary, and primary carbocation
character, respectively.
- It is important to note, as is also shown in the above diagram,
that a disubstituted alkene having both alkyl groups attached to the same
carbon, such as isobutene, reacts much more rapidly than a disubstituted alkene
having one alkyl group on each carbon, such as 2-butene, because positive
charge is developed upon only one of the alkene carbon atoms. Thus, the
TS for addition to 2-butene has only secondary carbocation character, the
second alkyl group not being at a site of positive charge and exerting little
effect upon the reaction rate.
CARBOCATIONS
- The important new point to be added here is that carbocations,
though extremely reactive as a class, vary greatly in their stability and
reactivity. In particular,the stability order is tertiary>secondary>primary>methyl.
The reactivity order is just the reverse. That is, the methyl carbocation
is the least stable and the most reactive. The classification of primary,
secondary, etc. is made on the basis of whether the trigonal, carbocation
center has 1,2, or 3 carbons bonded to it.
- The replacement of each hydrogen on the methyl carbocation
by an alkyl group furnishes an increment of additional stabilization. We say
that alkyl groups stabilize a carbocation center.
- The amount of stabilization per alkyl group is approximately
15-20 kcal/mol even in the solution phase. It is much greater than this
in the gas phase. The amount of the stabilization is thus relatively large,
either in solution or in the gas phase.
- The stabilization of a carbocation center by an alkyl group
occurs by two distinct means: (1) a resonance effect, specifically,
hyperconjugation and (2) an inductive effect.
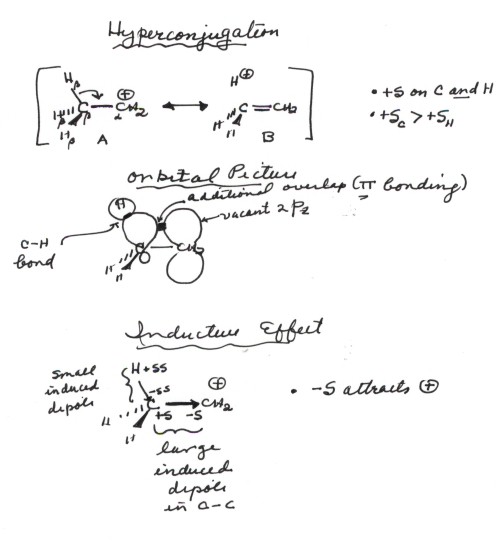
- Hyperconjugation. You recall
that a resonance effect refers to situations in which a single structure does
not adequately represent the bonding and charge distribution in a molecule.
The electrons are thus more highly delocalized than in any single canonical
structure. A hyperconjugative effect is a specific type of resonance effect
in which the delocalization involves a sigma bond. Referring to the resonance
structures for the ethyl carbocation, shown below, structure B is also a valid
structure which has the same number of bonds as that of A (although they are
of different types). Notice that B shows that the positive charge is not localized
completely upon the central carbon atom, but is delocalized onto the hydrogens
which are beta to the carbocation center. To put it differently, the electrons
in the C-H sigma bonds are delocalized onto the carbocation site, forming
a partial pi bond between the carbocation site and the attached carbon atoms.
The real structure of the ethyl carbocation is thus a resonance hybrid
of these two structures and is intermediate between the two. However,
since structure A is of lower energy than B, the real structure more closely
resembles A than B. Thus, there is more charge on carbon
than on H, and the carbocation tends to react more readily at carbon than
at H. Note that A is of lower energy than B because a relatively strong
C-H bond in A is replaced by a much weaker C-C pi bond in B.
- The overlap which gives rise to the resonance stabilization/delocalization
is between the 2p orbital on the carbocation center and the sp3
orbital on the beta carbon. If there is no beta carbon, as in the methyl carbocation,
this critical overlap is missing.
- Each additional alkyl group attached to the carbocation
center provides additional delocalization, so that the isopropyl carbocation(which
has two alkyl groups) is more stable than the ethyl carbocation(which has
just one) and the tertiary butyl carbocation(which has the maximum of three)
is more stable than either. Please note that the methyl carbocation has no
beta hydrogens to delocalize the charge. Alpha hydrogens can't delocalize
the charge, which you can verify by attempting to write good resonance structures
for the methyl carbocation (there are none).
- The inductive effect which stabilizes a carbocation
center results from the induction of a bond diple in the C-C and the C-H bonds
by the carbocation center. The positive charge on this carbon makes it very
electron deficient, so that it behaves like a very highly electronegative
atom and this makes the C-C bond very polar covalent bond, with the negative
end at the carobocation center. This partial negative charge is attracted
to and stabilizes the positive charge of the carbocation center(the negative
potential energy of attraction of opposite charges). Similarly, the positive
charge on the beta carbon, makes it somewhat more electronegative than is
usual for carbon, and it induces dipoles in the C-H bonds which are oriented
with the negative end of the dipole closer to the carbocation center, resulting
in a net stabilization. So the inductive effect here refers to the induction
of bond dipoles in the neighboring bonds.
REFINING THE TS MODEL: THE
HAMMOND PRINCIPLE
- Finally, it is important, in the consideration of TS models,
not only to be able to discern the qualitative character of the TS, but also
to have some idea of the extent to which this character is developed
in the TS.For example, in the purely hypothetical case that the addition of
HCl to an alkene had only a very minor amount of carbocation character upon
the passive carbon, the rates of reaction of isobutene, propene, and ethene
might not be very different at all. Further, the rates of protonation of the
two non-equivalent carbons of isobutene might not be very different, so that
both product regioisomers would be produced in significant amounts. The reaction
would thus be said to be relatively "unselective". If,
however, there is very extensive development of carbocation character, so
that the TS closely resembles a carbocation, we can expect that the reaction
will be very selective. Rate differences between various alkenes will
therefore be very large, and the regiospecificity of addition to an unsymmetrical
alkene will also be high.
- The Hammond Principle prodvides an excellent approach
to further refining our TS as to the extent of a given character developed
in a TS. Essentially, the postulate states that "The
TS of a given reaction step more closely resembles the reaction partner (reactant
or product) of highest energy." Thus, if a reaction is highly
endothermic (or endergonic), the TS tends to more closely resemble
the product than the reactant, although it is still a resonance hybride
of both. If, on the other hand, a reaction is highly exothermic (or exergonic),
the TS tends to more closely resemble the reactant. Traditionally,
the Hammond Principle is discussed in terms of enthalpies, that is, endothermicity
and exothermicity.
- It is important to note that the Hammond Principle helps
define the TS for a specific, single reaction step, not the overall reaction.
Since we are considering relative rates, the step we are most interested in
is the rds, becasuse the rate of the reaction equals the rate of this step.
In the addition of HCl to an alkene, this step is quite endothermic. We could,
and would be expected to, simply look at this reaction step and predict that
it would be endothermic. Remember that two bonds are broken in this reaction
and only one formed. It is thus substantially endothermic. As a matter of
convenience, it may be well to remember that any time a high energy intermediate
such as a carbocation is formed, that reaction step is endothermic.
From the Hammond Principle we may then conclude that the
TS for this step closely resembles the product (of that
step), which is a carbocation. Therefore the TS for the rds of
this reaction has very extensive carbocation character.
- On the other hand, the final step (which is fast and
not rate determining) is highly exothermic, so the TS for that step (reaction
of a carbocation with the chloride anion) closely resembles the reactant.
By the way, the reactant in this step is also this same carbocation. Therefore
the TS for both steps closely resembles the carbocation.
Nevertheless, we are presently interested only in the rds (because we are
concerned with relative rates).
- Consequently, we can predict that rate
ratios in the series of alkenes previously considered will be large, and also
regiospecificity in the addition to an unsymmetrical alkene will be high,
as indeed is the case.
BACK TO THE TOP OF THIS PAGE
BACK TO ALKENES 1
ON TO THE FINAL PART OF ALKENES
BACK TO THE BAULD HOME PAGE